HERITABILITY
Psychiatric syndromes have long been known to be familial. Demonstrating heritability involves three types of studies: family, twin, and adoption studies. Family studies are used to establish family aggregation and also to make inferences regarding the heritability of a disease or trait. This design involves using the presence of a phenotype in relatives of varying degrees and examining this in a segregation analysis, which can help define the nature of the model of inheritance. Once heritability is established, the task becomes teasing out the relative contributions of environment and genes.
Twin studies help assess genetic influences in the context of a shared environment. They compare concordance rates between monozygotic (identical) twins, who share nearly 100% of their genes and dizygotic (fraternal) twins, who share on average 50% of their genes (same as ordinary siblings). An expectation of shared environment is made because both types of twins mature together in the uterus and often grow up together in the same home. Differences in concordance rates between monozygotic and dizygotic twins help define the degree of genetic versus environmental risk (
Table 1). The patterns seen in both schizophrenia and bipolar disorder are consistent with a strong genetic risk. In 1971 Fischer (
4), in a study of the children of monozygotic twins with schizophrenia, showed that the offspring of the unaffected co-twin carried the same risk as the children of the affected co-twin. Clearly, genetic risk carried by a parent is transmitted to a child, whether the parent expresses the illness or not. However, the incomplete concordance in monozygotic twins provides evidence for nongenetic or environmental risk. The term
penetrance is used to connote the proportion of individuals with a disease-causing gene(s) who exhibit clinical symptoms or other phenotype of interest. Nonconcordant monozygotic twins suggest incomplete penetrance of the disease-causing gene(s).
Other approaches to teasing apart environmental from genetic influences are adoption and cross-fostering study designs. One can look at a patient's biological and adoptive families and look for familial aggregation. In a series of landmark studies in the 1960s by Heston (
5), Kety et al. (
6), Wender et al. (
7), and others, the adopted away children of mothers with schizophrenia were found to have an increased risk of schizophrenia despite having been raised from birth by nonschizophrenic adoptive parents. Furthermore, children adopted at birth by adoptive families segregating for schizophrenia showed no increased risk for the illness. In a genetically determined disease, one should find exactly these results: higher rates of illness only in the biological family.
GENETIC EPIDEMIOLOGY
Once a genetic contribution to disease is demonstrated through family, twin, and adoption studies, both genetic epidemiological and molecular genetics approaches may be applied to identify causative gene(s). Genetic epidemiological studies seek to identify not only the gene or genes involved in disease etiology but also the interaction of these genes with environmental factors that may range from the in utero environment to the social and psychological environment to the cellular environment. Even if a putative causative gene is known, the pathway from gene to the development of disease or an observable characteristic (“phenotype”) may be complex, depending on the gene's penetrance, the influence of other genes (“epistasis”), and the influence of the environment. In recent years, the importance of the cellular environment of the gene—the architecture and biochemical milieu surrounding and supporting the DNA molecules—has been recognized. Changes in this environment are heritable and can affect expression of the gene product and may result in particular phenotypes. Changes in gene expression or phenotype that are due to mechanisms other than alterations in the underlying genes are known collectively as epigenetics, whereas the term epigenome refers to the overall epigenetic state of a cell.
In many medical disorders, especially psychiatric disorders, life events (stressors) should be considered as part of the environmental risk. This consideration fits with a diathesis-stress model, which postulates that interaction of environmental factors, including stressful life events, with an underlying susceptible biological or genetic profile can cause the emergence of particular behaviors or other phenotypes. There is an inverse relationship between the underlying vulnerability (diathesis) and the environment: the greater the vulnerability, the less the stress need be to evoke the behavior or other phenotype. This result implies that a threshold effect may be occurring.
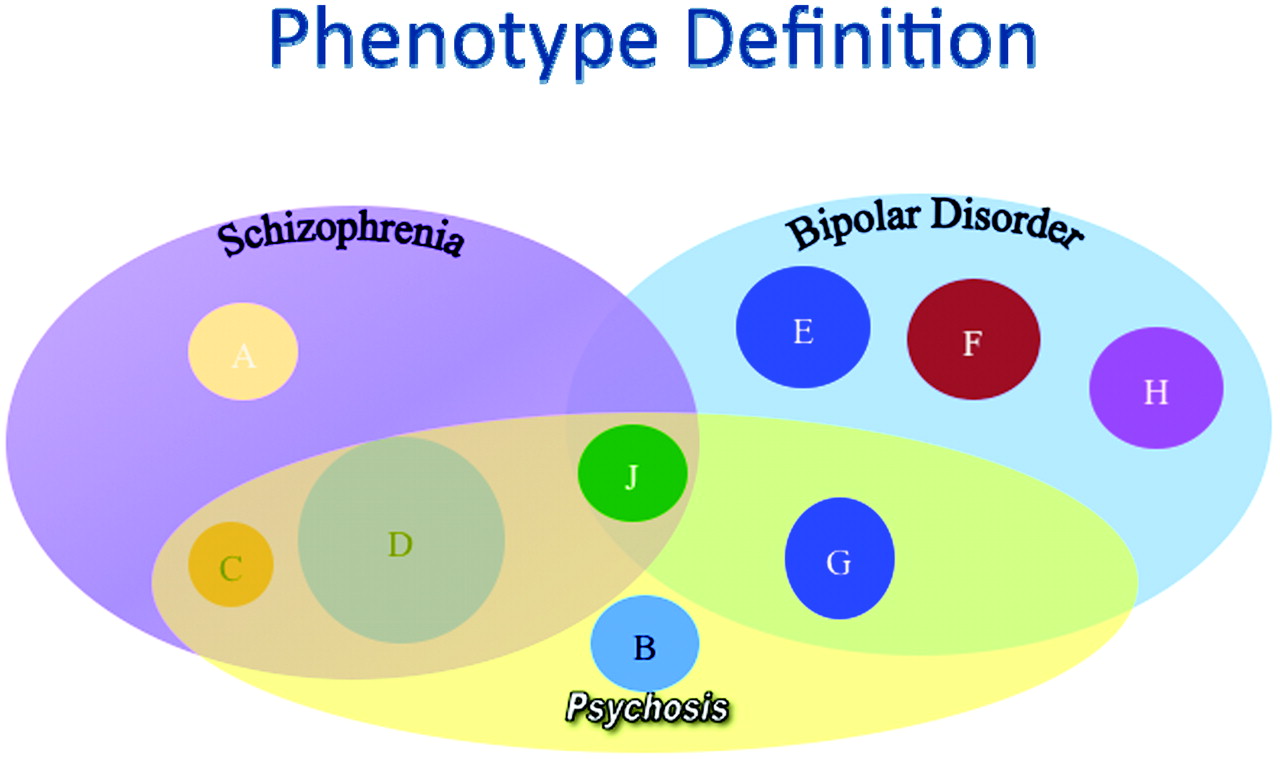
In addition, psychiatric phenotypes, such as schizophrenia, are syndromes that may include a variety of subtypes with different specific etiologies but with a shared clinical presentation. We know that bipolar disorder and schizophrenia are distinguishable clinically although they may share a number of symptoms (i.e., psychosis). It is equally possible that these two complex syndromes may share some of the same genetic underpinnings as well as clearly distinct risk factors. These complex issues have made understanding the genetics of these disorders more complex (
Figure 1).
MOLECULAR GENETICS
The genetic code of DNA is made up of four nucleotide bases (adenine, guanine, thymine, and cytosine) that are arranged linearly along DNA molecules into triplet reading frames, called codons, that are “translated” into the gene's product. There are roughly 3 billion base pairs in the human genome. The estimated 22,000 human genes are packaged into 22 pairs of maternally and paternally derived chromosomes, plus a pair of sex chromosomes (XX or XY, respectively). The size of a gene may range from a few thousand to more than a million base pairs. Regulatory sequences are built into the gene structure, such as binding, promoter, initiation, and enhancer sites, that are integral to the control of gene expression. In differentiated cells, most of the genes are turned off.
The DNA is transcribed into RNA within the nucleus and translated into protein in the cytoplasm. Long stretches of noncoding regions within the gene, called introns, are transcribed into the initial mRNA molecule but are spliced out of the final transcript. It is estimated that 98% of the human genome is made up of noncoding sequences, of which introns are just one type. Much of this noncoding sequence is, in fact, coding for RNA that, in turn, is functional on its own. We continue to uncover a far more complex genomics that lies beyond our traditional understanding of genes.
MUTATION AND POLYMORPHISM
The process of mutation, defined as changes in DNA sequence, is not always deleterious—it has allowed organisms to adapt to a changing environment and is responsible for the genetic variation in a population. The overwhelming majority of sequence changes in the human genome are of no functional consequence. These DNA variations occur at single nucleotide bases (e.g., an adenine nucleotide is changed to a guanine nucleotide). Approximately one of every 1000–1200 base pairs of DNA differ among individuals and result in slightly different forms of the gene (alleles). Alleles at different loci along a chromosome that are cotransmitted, or inherited together, are known as a haplotype.
If the allele at a locus (chromosomal location) occurs at least in 1% of the population, it is considered polymorphic (i.e., many forms). The abundant DNA sequence variants at single nucleotides are known as single nucleotide polymorphisms or SNPs (pronounced “snips”) because, by definition, they occur in at least 1% of the population. Sequence changes that occur in less than 1% are termed rare variants. SNPs are heritable, and when the alleles they constitute at different loci along a chromosome are cotransmitted, they are known as a SNP haplotype.
SNPs have become powerful markers for the conduct of genetic linkage and association studies. Through the International HapMap Project, SNPs have been mapped along the length of every human chromosome for use in disease-associated gene mapping. SNPs coordinately inherited can be identified as specific haplotypes and the frequency of both individual SNPs as well as SNP-based haplotypes can be assessed in individuals with disease (case subjects) and those without (control subjects). SNPs and/or haplotypes with a statistically significant increased frequency in case subjects may be presumed to be associated or, if transmitted within a family, coinherited with an unknown disease-causing mutation. Case-control association studies have become a popular method, in addition to pedigree-based linkage analysis, to identify disease genes. Highly automated multiplex assays may be performed to simultaneously analyze a million or more SNPs throughout the genome. Genome-wide association studies (GWASs) may be performed in case subjects and unrelated control subjects (or between affected and unaffected family members) to determine which SNPs segregate with a disease, trait, or other phenotype of interest. Because SNPs are already mapped to specific chromosomal locations, the location of the putative disease-associated gene is known (although this may be thousands, if not more, base pairs away). The exact identification of a causative mutation and replicated demonstration of disease causality remain to be established.
LINKAGE ANALYSIS
Individual SNPs and SNP haplotypes may be used as “markers” in families segregating disease to identify an unknown disease susceptibility gene or chromosomal region that may harbor a susceptibility gene(s). By observing the coinheritance of disease with particular markers in the affected and unaffected members of families, the existence of a linked deleterious gene may be determined.
Genes and markers (such as SNPs or SNP haplotypes) in close proximity to each other on the same chromosome are usually cotransmitted as single units known as linkage groups. If the sequences are further apart on the chromosome, they are more likely to be separated by recombination events between paired homologous chromosomes during meiosis. The greater the distance between two loci is, the more physical space that exists for this “crossing over” process to occur, resulting in a shuffling of alleles between maternally and paternally derived chromosomes. Thus, alleles at two physically close loci tend to be transmitted to offspring in the same combinations in which they were received in the sperm or egg, whereas those on different chromosomes or far apart on the same chromosome are transmitted independently. Because the recombination frequency along chromosomes is mathematically predictable, distances between markers and genes can be estimated by counting the number of recombinants occurring within families or pedigrees. A genetic map can be constructed using mapping units that correspond to recombination frequencies. A map unit of 1 centimorgan (abbreviated cM) signifies a 1% chance that a marker at one genetic locus will be separated from another locus by a recombination event in a single generation. In the human genome, 1 cM is equal to 1,000,000 base pairs [1 megabase (Mb)] on average, although there is variability in recombination frequencies among the chromosomes and between the female and male genomes.
In linkage analysis, an unknown, but presumably causative, genetic variant may be localized proximal to a marker of known location. A statistical test is performed that compares the likelihood that the observed pattern of coinheritance would have resulted if the marker and the unknown gene were truly linked with the likelihood that the observed data were due to chance. This statistic is known as the logarithm (base 10) of the odds (LOD) score. Traditionally, a LOD score greater than +3 (1000:1 odds) has been considered evidence in favor of linkage. Both parametric (underlying model of inheritance, such as autosomal dominant, is specified) and nonparametric (no underlying model of inheritance specified) statistical analyses may be performed (depending on the nature of the study sample and other considerations).
COMPLEXITIES OF DISEASE-GENE MAPPING IN PSYCHIATRY
Both linkage analysis and GWASs hold promise for identification of the underlying genetic causes of the major psychiatric disorders, including autism, schizophrenia, and bipolar disorder. Although reasonably straightforward in theory, in practice the approaches present difficulties that are inherent to the diseases and to the human populations in which they occur. As described earlier, disease definition is often syndromal, and there may be phenotypic overlap in some symptoms (e.g., psychosis in schizophrenia and bipolar disorder or autistic features in many distinct disorders) or unclear diagnosis in others (e.g., psychosis not otherwise specified). Although autism, schizophrenia, and bipolar disorder have been shown to have high heritability, the existence of a single causative gene for each disorder is unlikely. Rather, each of these disorders is presumed to involve many genes (polygenic) and also to involve environmental risk factors. Collectively, disorders with these attributes are referred to as multifactorial. Even if the underlying genes and environmental components were identified for a particular disorder, the exact combination of genes and/or environmental factors would probably differ among different population groups (e.g., African-Americans and Asians) or subgroups (e.g., Caucasians of Ashkenazi Jewish descent) as a result of how different populations developed and diverged over evolutionary time.
Because of these inherent difficulties in psychiatric genetics, researchers not only strive to study clearly defined cases of disease but also to develop study samples from relatively homogeneous population groups. We focused our work over the last 20 years on this strategy defining the Portuguese Island Collection studies. In brief, individuals with either schizophrenia or bipolar disorder, as defined by the DSM-IV criteria, were ascertained on the geographically isolated and genetically homogeneous Azores and Madeiran islands off the coast of Portugal (
8). These once unpopulated islands were settled more than 500 years ago almost exclusively by the Portuguese.
LINKAGE ANALYSIS IN THE PORTUGUESE ISLAND COHORT (PIC)
Schizophrenia
Our research group, in collaboration with researchers at the Broad Institute, identified a region on chromosome 5q31–5q35 with a nonparametric linkage score (NPL) of 3.28 in the PIC sample (
9). The finding was replicated in the PIC population with bipolar disorder with psychosis (
9). Within the region, SNP associations were demonstrated with several γ-aminobutyric acid (GABA) receptor subunit genes in both Portuguese and German patients with schizophrenia (
10). Other investigators have reported associations with the Epsin 4 gene on 5q33.3 (
11–
13). The gene codes for enthoprotin and may be involved in the transport and stabilization of neurotransmitter vesicles. In a meta-analysis using linkage data across multiple studies in Caucasian individuals with schizophrenia, chromosomal region 5q23.2–q34 was ranked second in strength of the evidence for an association with schizophrenia (
14).
Further collaborative efforts were undertaken using European Caucasian samples that had shown linkage within 20 cM of the region of linkage in the Portuguese samples. The combined samples had maximum evidence for linkage approximately 1.8 cM from perhaps the strongest candidate genes in the region, the GABA A receptor cluster. The relative proximity of the linkage in each sample is encouraging, and the region in which putative causative genes might reside has been shortened. A second, smaller peak at approximately 130–139 cM contributed to by both the Portuguese and German samples was noted.
Bipolar disorder
An initial genome scan was performed in 16 PIC extended families segregating for bipolar disorder. Regions on chromosomes 2, 11, and 19 showed suggestive evidence of linkage, and regions on chromosome 6q were just under the levels suggestive of linkage. In a follow-up study in the PIC, additional data supported linkage on chromosome 6q with a peak NPL=2.02 (p=0.025) with marker D6S1021 (104.7 Mb). Using a higher density of markers (10,000 SNPs), additional support for linkage to loci on chromosome 6 was found, with the strongest support being at marker D6S1639 (125 Mb) with NPL=3.06 (p=0.0019). Likewise, the initial finding of suggestive linkage on chromosome 11 also was strengthened in the denser scan [NPL=3.15, p=0.0014 at marker D11S1883 (63.1 Mb)].
Combining data from 11 independent linkage studies of bipolar disorder, including our PIC study, a collaborative group found strong evidence for linkage on chromosomes 6q and 8q. In total, genotype data on 5,179 individuals from 1,067 families were analyzed using novel statistical methods for meta-analyses and a common, standardized marker map (
15). Although only 400 markers on average were available across the studies for analysis, the chromosome 6 linkage was confirmed with NPL>3. The samples contributing most to the observed linkage derived from the PIC and two other groups (Columbia and National Institute of Mental Health Wave 3).
COPY NUMBER VARIANTS (CNVS)
CNVs occur when there is a deletion or duplication of a piece of a chromosome that changes the number of copies at a locus from the usual two copies to a different number of copies. CNVs have been found in patients with schizophrenia (
16), but they have also been found in patients with other neurodevelopmental disorders (
17–
19). We are part of the International Schizophrenia Consortium (ISC) and as part of this collaborative we have investigated possible CNVs in schizophrenia.
One of the known chromosomal anomalies related to psychosis is known as the “22q11.2 deletion syndrome” (22q11.2DS) (
20). Deletions in this region include the velo-cardio facial syndrome (VCFS) and DiGeorge syndrome. Approximately 30% of deletion carriers develop psychosis (
20) and VCFS deletions occur in 0.6%–2.0% of individuals with schizophrenia (
21,
22). Based on this observation, we had a strong prior expectation that 22q11.2 deletions would be identified. Among the ISC sample of more than 6,500 individuals, 13 individuals with schizophrenia were found to have large deletions (i.e., > 500 kilobases) in the 22q11.2 region (
23). None of the control subjects showed deletions. Eleven of the 13 schizophrenic individuals had very large deletions (>3 Mb) which is the most common form found in VCFS carriers. In an analysis restricted to this deletion form, the odds ratio was 21.6 (empiric p=0.0017; genome-wide corrected p=0.0046) (
23).
In addition to the 22q11.2 deletions, large deletions were identified on chromosomes 15q13.3 and 1q21.1 (
23). The 15q13.3 deletion (28–31 Mb) was observed in nine case subjects but in no control subjects (empiric p=0.0029; genome-wide corrected p=0.046; odds ratio=17.9) (
23). A smaller but significantly elevated odds ratio (6.6) was found for the 1q21.1 deletion (142.5–145.5 Mb). Ten case subjects and one control subject carried deletions (empiric p=0.0076; genome-wide corrected p=0.046) (
23). In total, large deletions at these three loci occurred in nearly 1% of the individuals with schizophrenia. These results were replicated by others in a concurrently published report (
24).
BRIEF DESCRIPTION OF SCHIZOPHRENIA DELETION REGIONS
Chromosome 15q13.2
The 15q13.2 deletion is distinct from the region disrupted in Prader-Willi/Angelman syndrome (
25) but does incorporate deletion regions that have been observed in individuals with mental retardation and seizures (
26). Among the nine individuals with schizophrenia who have this deletion, five had a history of mildly impaired cognition and one had an additional history of epilepsy. The deleted region contains the α7 subunit of the nicotinic acetylcholine receptor (
CHRNA7). This gene has been implicated in linkage analysis of the P50 auditory evoked potential deficits observed in patients with schizophrenia (
23,
27,
28).
Chromosome 1q21.1
The deleted region on 1q21 is consistent with a report on two patients with autism (
19). In the ISC study, mild cognitive abnormalities were noted in three patients, and there was a history of epilepsy in another (
23). The deleted region encompasses 27 known genes. Linkage in this region has been reported by others (
29,
30).
Chromosome 22q11.2
The larger 22q11.2 deletion region encompasses 43 genes, several of which have been reported as being associated with schizophrenia. The most widely studied is the catechol-
O-methyltransferase (
COMT) enzyme involved in the degradation of dopamine and other catecholamines.
COMT variants have been implicated in a wide variety of phenotypes, but the findings have not been consistently replicated (
20,
31).
POLYGENES WITH SHARED EFFECTS BETWEEN SCHIZOPHRENIA AND BIPOLAR DISORDER
A larger GWAS was conducted by the ISC and included 3,322 European case subjects with schizophrenia and 3,587 control subjects (
32). More than 1 million SNPs were assayed. This sample had been the basis for our original report on CNVs. The major histocompatibility complex on chromosome 6p was implicated. Based on statistical genetics models, the authors estimated that common polygenic variations of small individual effect could comprise at least one-third of the total variation in schizophrenia risk. These findings again were replicated in a number of other reports published concurrently (
33,
34).
Furthermore, when one examines the subset of common variants that showed nominal association with disease and compared these among large samples of patients with schizophrenia, there is an extremely high degree of concordance between samples. Interestingly, in the same type of analysis using populations studied for other disorders (coronary artery disease, Crohn's disease, hypertension, rheumatoid arthritis, and type I and type II diabetes), the groups were entirely unrelated. In contrast, strong statistically significant associations were detected in populations of patients with bipolar disorder. This finding may provide evidence for shared genetic risk among these two syndromes (
32).
SUMMARY OF CONTRIBUTIONS TO THIS ISSUE
The new articles in this issue of FOCUS explore some recent developments in psychiatric genetics and genomics. The articles presented here discuss both the advances in the field and challenges being met across the field.
Campbell (
35) discusses the technological and methodological advances in autism spectrum disorder (ASD) research and the future directions of the field. As in a number of other disorders, the approaches being explored include larger sample size linkage studies, GWASs, and analysis of CNV to identify deletions and duplications. Campbell discusses how recent studies using these methods have offered support to candidate genes including
MET, GABRB3, EN2, SLC6A4, and
OXTR. As we have commonly seen, GWASs have not replicated these results but have identified new regions of interest within the genome. These findings warrant further exploration and continued technological advancement in both sequencing techniques and CNV platforms will help greatly.
Campbell also highlights two major issues that may offer further insight: the observation that ASD affects four times as many males as females and that, because there is no single strong candidate gene, it is likely that multiple genes contribute to ASD risk. The ultimate challenge of the field is to now understand how the genetics affect brain development and to determine effective forms of treatment.
Fanous (
36) discusses recent findings on the genetics of schizophrenia and bipolar disorder with particular attention paid to the genetic relationship between the two disorders. His review of the literature showed that although large samples sizes and modern methods have yielded many interesting findings, there are only a few sequence variations that have been replicated. These include SNPs in
ZNF804A, MHC genes, and
CACNA1C and deletions in 1q21.1 and 15q13.3. In many of these cases, there is evidence for association in both schizophrenia and bipolar disorder, which supports a possible genetic relationship between these syndromes.
Fanous goes on to discuss how this overlap is probably due in part to the current diagnostic criteria for schizophrenia and bipolar disorder and how studies chose to group subjects. He proposes that future researchers examine more specific phenotypes such as removing patients with bipolar disorder with psychosis and affective symptoms of patients with schizophrenia from analyses. He also stresses the importance of continuing to use new modalities to delve deeper into the underlying explanations for these associations. Further, other factors that may play a role such as environmental variables, gene-gene interactions, gene-environment interactions, the interaction between sequence and epigenetic variation, and the limitations of current technology must be taken into account.
Hettema (
37) examines current large-scale studies and GWASs and how their findings are affecting our understanding of the genetics of major depressive disorder (MDD). MDD is characterized by frequent comorbidities with other disorders such as anxiety disorders. An individual's susceptibility to MDD is probably due to a combination of factors including the interaction of multiple genes and life events. To detect these interactions and clearly distinguish these disorders it is becoming increasingly necessary to use larger samples and more specific phenotypes to understand the genetics of MDD.
Recent GWASs using large samples and modern high-throughput genotyping technology have yielded promising results. Interestingly, these GWASs did not replicate the findings of prior association studies but did suggest two novel susceptibility candidates, GRM7 and PCLO. There are a number of more powerful GWASs underway that may supply further understanding of the genes involved in MDD susceptibility. Further exploration into how life events and genetics interact to cause illness in some but not others should greatly increase our understanding of MDD.
Stewart and Pauls (
38) review recent studies on the genetics of obsessive compulsive disorder (OCD). In the past few decades, a number of linkage and candidate gene studies in OCD have identified a variety of possible candidates, but it has been difficult to replicate the findings. The only candidate gene that is replicated across studies is the glutamate transporter gene,
SLCL1A1.
Stewart and Pauls believe that this lack of replicable findings is probably due to the fact that risk for OCD is caused by small effects from many genes and that the next logical step in the field is to invest greater effort in GWASs. GWASs provide far more power when identifying risk genes with small effects. They cite one GWAS currently being conducted by the International OCD Foundation Genetics Collaborative and believe that the findings of this study will greatly forward our understanding of the genetics of OCD.
Deng et al. (
39) introduce the terms and concepts of epigenetics and reviews recent data suggesting the influence of epigenetic alterations in schizophrenia. As noted earlier, epigenetics broadly refers to heritable changes in the expression of a gene or in a phenotype that are not due to the underlying DNA sequence but rather to other mechanisms. The epidemiology of schizophrenia, including the less than 100% concordance patterns in monozygotic twins, the peak age of onset in late adolescence and early adulthood, the variability in age of onset between males and females, and the reported associations with in utero nutritional deficiency, viral exposure, and hypoxia together suggest that epigenetic factors may be important in disease etiology.
Deng et al. describe the major epigenetic mechanisms of DNA methylation, genomic imprinting, histone modifications, and expression control by noncoding RNA. They highlight recent human and animal model literature suggesting epigenetic aberrations in schizophrenia and schizophrenia-like behaviors.
With the field of psychiatric genetics growing rapidly, it is crucial to consider the ethical implications of clinical genetics as it pertains to psychiatric disorders. Hoop (
40) examines the four primary ethical issues in genetic testing: testing may predict a person's future health, it may have psychosocial consequences, it affects family members and communities, and it is a rapidly evolving field. She reviews the existing literature, exploring these issues within psychiatric genetics.
The findings of these studies are in agreement that patients and their families are interested in the clinical applications of psychiatric genetics. However, limitations in the study designs have made it difficult for them to explore the issues mentioned above. Hoop stresses the urgent need to design protocols to directly address these issues and the issues posed by psychiatric genetic research. As the field continues to evolve, constant reflection on the ethical issues surrounding psychiatric genetics is essential to protect patients and supply them with the best care possible.
CONCLUSION
This issue of FOCUS was designed to both introduce the reader to the advances in psychiatric genomics and help guide the practitioner by explaining the methods and terms used in this field of research. The complexity of the genetics of most human disease and certainly most forms of psychiatric disorders does present a challenge. However, rapid technological advances continue to make this field of research one with the greatest potential to help us understand risk, etiology, and potential new treatments.