Neuroimaging.
Functional neuroimaging studies have identified abnormalities in the brains of individuals with BPD, which may be indicative of dysfunction in key neural circuits (
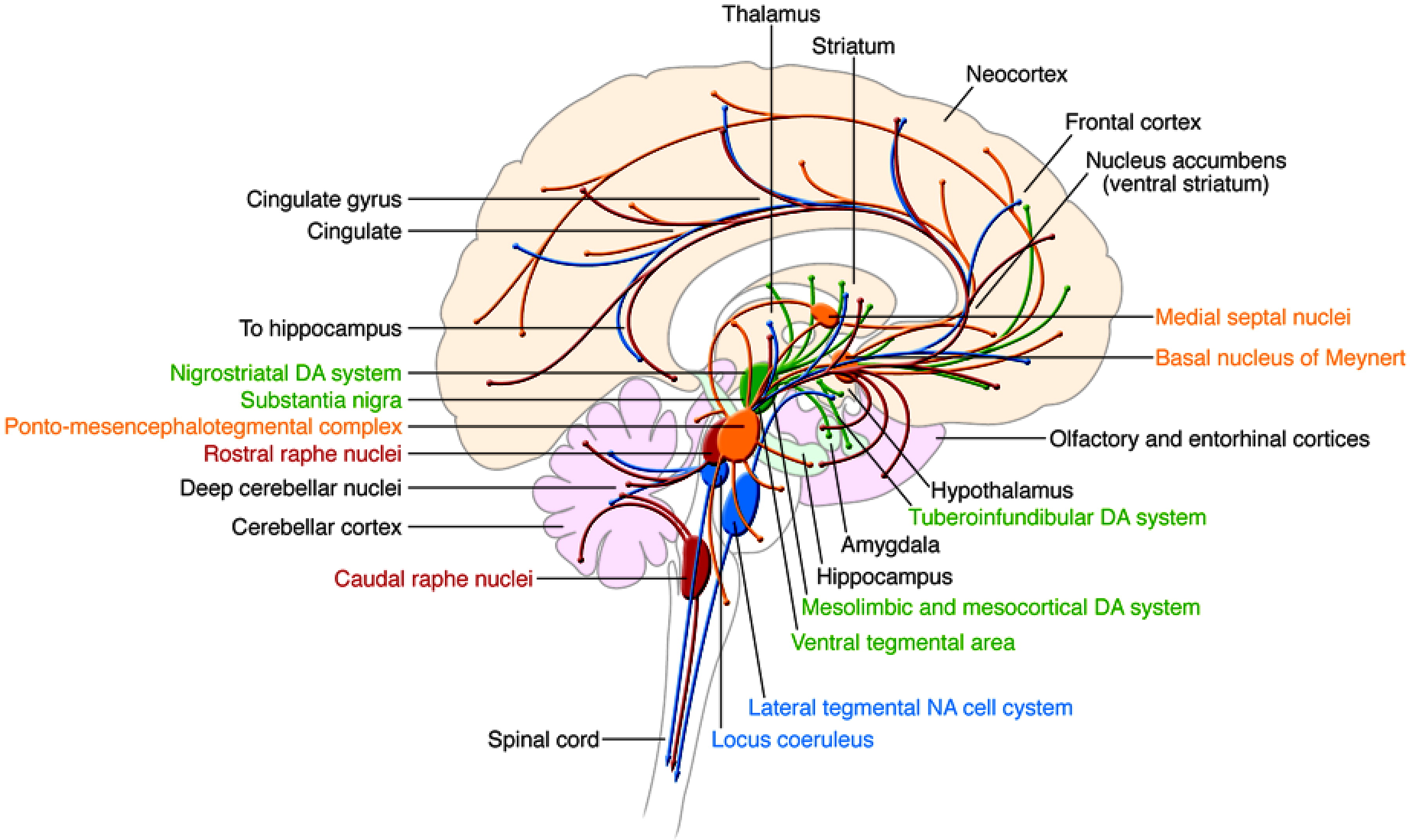
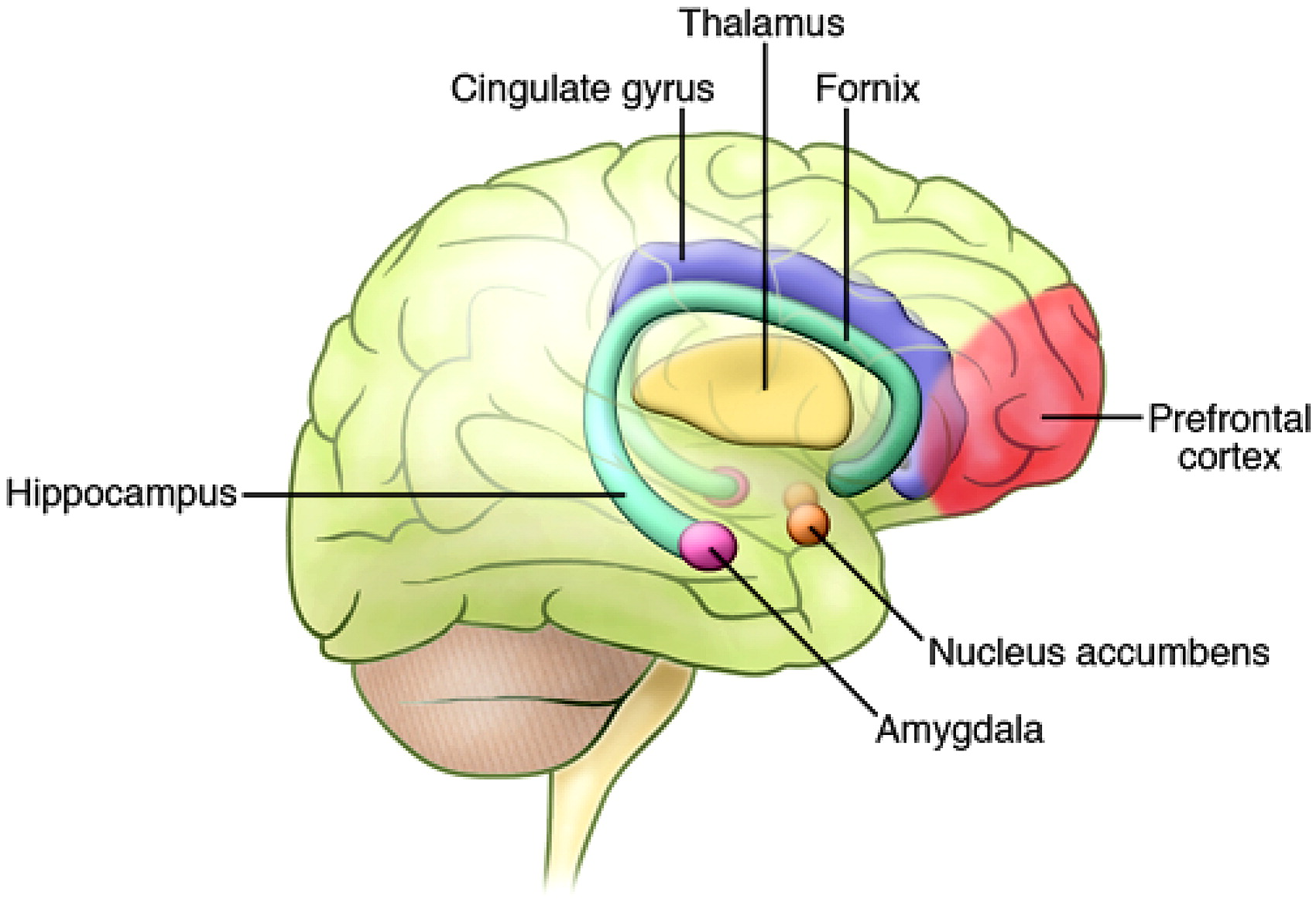
4). These circuits are distributed throughout a wide array of neuronal structures, including the amygdala and related limbic nuclei, the orbital and medial prefrontal cortex (PFC), the anterior cingulate, the medial thalamus, and related regions of the basal ganglia (
Figure 2) (
5). It is plausible that dysfunction or modulation of these circuits may predispose an individual toward manifestation of the symptoms of BPD. For example, studies have shown reduced activity in the right PFC during episodes of mania (
6), and dysfunction of the right PFC is thought to contribute to a disinhibited profile, including poor impulse control, risk taking, distractibility, poor sustained attention, and delusions, all of which resemble the symptoms of mania (
4,
7).
Researchers using CT and MRI have identified structural changes in the brains of patients with mood disorders, including patients with BPD. Overall, gray matter volume is not substantially different in patients with BPD compared with normal healthy individuals (
8–
10). However, several studies have found region-specific reductions in gray matter volume, including increased ventricular size and decreased frontal cortical volume (
11,
12). Specifically, volumetric decreases have been identified in the anterior cingulate cortex (
13), and other studies have indicated gray matter loss in the left dorsolateral PFC (DL-PFC) (
14), the ventral PFC, and the orbital PFC (
15). Temporal lobe regions, including the hippocampus and the amygdala, have not been as well studied. However, one study found more prevalent volumetric decreases in the right hippocampal formation of the affected twin in monozygotic twin sets discordant for BPD (
16).
White matter hyperintensities (WMHs) around the ventricles and in the subcortical white matter have consistently been found in the brains of depressed elderly persons as well as in the brains of patients with BPD (
17,
18). Although the functional and pathological relevance of WMHs has not been fully determined, WMHs have been associated with cerebrovascular accidents (stroke and transient ischemic attacks), ischemia, axonal loss, increased perivascular space, minute brain cysts, and necrosis (
19). Recent data suggest that a substantial proportion of individuals with BPD, including children diagnosed with BPD, exhibit WMHs more frequently than the general population. The incidence of WMHs is higher in children with neuropsychiatric disorders, including children with BPD (
20,
21). In addition, WMHs are associated with poor treatment outcome in patients with mood disorders, particularly when localized to the subcortical rather than the periventricular areas (
22,
23). Combined studies on the increased incidence of WMHs in affective disorders suggest that these lesions could indicate some type of damage to brain tissue, which could result in disruption of the neuronal connections necessary for normal behavioral functioning (
24).
Imaging studies using magnetic resonance spectroscopy (MRS) led to the notion that BPD may be associated with mitochondrial dysfunction (
25,
26). In particular, high-resolution
1H-MRS imaging studies conducted to quantitatively assess concentrations of
N-acetyl-aspartate (NAA), a predominant neurochemical in the human brain that is localized to mature neurons and synthesized within mitochondria, found decreased concentrations of NAA in the hippocampus, the DL-PFC, the orbitofrontal cortex, and the basal ganglia in various patient populations, including patients with BPD (
27–
32). In addition, studies using
31P-MRS, which allows examination of energy metabolism in the brain, showed a decrease in phosphocreatine and/or ATP levels in patients with a mood disorder, including patients with BPD (
33–
35). Consistent with these findings, low brain pH levels, as measured indirectly by
31P-MRS, have also been identified in patients with a mood disorder (
33–
35).
While it is clear that BPD is not a mitochondrial disorder per se, some data suggest that cellular and molecular abnormalities present in the brain of patients with BPD may be associated with alterations in normal mitochondrial function (
36). Evidence from microarray, biochemical, neuroimaging, and postmortem studies support the role of mitochondrial dysfunction in BPD (
37). In addition to energy production, neuronal mitochondria play an important role in regulating apoptosis, intracellular calcium levels, and synaptic plasticity (
24). Mitochondrial dysfunction may also be involved in the calcium signaling abnormality found in BPD (
37). Regulation of intracellular calcium levels may be particularly important in the context of the CNS, since fast changes in the levels of calcium are responsible for mediating actions associated with the release of, and response to, neurotransmitters. Changes in calcium levels are also crucial for initiating transcription events at several genes whose transcription is dependent on increases in neuronal activity. One of these neuronal activity-dependent genes, brain-derived neurotrophic factor (
BDNF), has been implicated in the etiology of several mood disorders (
38). Furthermore, it is known that mitochondria are involved in the initiation of apoptotic processes (
39), and newer evidence has suggested that proper mitochondrial function may be important for the regulation of synaptic plasticity. For example, increased neural activity has been shown to induce transcription of genes encoded by mitochondrial DNA, suggesting that increases in energy production may play a role in the regulation of synaptic strength (
40).
A role for the glutamatergic system in mood disorders, including BPD, has also been supported by neuroimaging data. In a proton MRS study of children with BPD, patients were shown to have increased levels of glutamate in the frontal lobes and basal ganglia (
41), while another study identified increased glutamate levels in the occipital cortex of depressed adult patients (
42). Although current data from neuroimaging studies are interesting, their interpretation remains incomplete and often controversial. Despite finding differences in specific locations and deficits in biochemical markers, it is not yet understood what these findings represent or how they may affect the function of various circuits in the brain. In addition, it is not yet known whether the abnormalities that have been discovered represent developmental problems that confer vulnerability to severe mood disorders, compensatory mechanisms for other related pathogenetic processes, or the continual recurrence of affective episodes.
Analysis of postmortem human tissue.
Studies of postmortem brain tissue from patients with recurrent mood disorders, including some with BPD, have found reduced subcortical nuclei volumes (
43). Various other studies have shown differential neuronal densities and morphologies that appear to be layer and cell-type specific in both patients with BPD and patients with major depressive disorder. For example, the density of neurons containing large cell somas, which are likely to correspond to glutamatergic pyramidal neurons, was decreased in layers III and V of the DL-PFC of individuals with recurrent mood disorders (
44). Furthermore, the size of DL-PFC neurons in layers V and VI was also reduced (
45). In patients with BPD, the size of the cell soma of pyramidal neurons in the CA1 region of the hippocampus has been found to be decreased (
46), as have cell densities in layers III–VI in different regions of the anterior cingulate (
24,
36). Interestingly, cell densities of nonpyramidal neurons were decreased in layer II, and neuron size was increased in layers II and V of these anterior cingulate regions (
24,
36).
In addition, decreased levels of calbindin and parvalbumin-expressing neurons, both of which are subtypes of GABAergic interneurons, have been identified in the anterior cingulate cortex, the hippocampus, and the entorhinal cortex of patients with BPD (
47,
48). These data, in combination with studies showing decreased hippocampal expression of glutamic acid decarboxylase 67 (GAD67) and somatostatin in the hippocampus of patients with BPD, have led to the hypothesis that a subset of hippocampal interneurons may be abnormal in BPD (
49).
In addition to changes in the size and density of neuronal cell types, changes in glial cell biology have also been observed in studies of postmortem tissue from patients with BPD. The density of glial cells seems to be decreased in frontal cortical areas, while the nucleus size increases (
44,
50–
52). Moreover, a proteomics study using brains from individuals with BPD and individuals with major depressive disorder found disease-specific alterations in levels of glial-fibrillary acidic protein (GFAP), an abundantly expressed astrocyte-specific protein (
53). Additional studies have found reductions in oligodendrocyte number and in the expression of genes that are related to oligodendrocyte differentiation and myelin production in the DL-PFC of individuals with BPD (
54). The recurrent theme of decreased cell density and number may represent cell loss and atrophy in these patients over the course of disease progression. It is presently unknown whether this type of brain atrophy is one of the underlying causes of the disease, or whether it contributes to illness pathology by disrupting the normal circuitry that is key to normal affective and cognitive functioning.
As discussed above, neuroimaging studies have provided suggestive evidence for abnormalities in mitochondrial function and energy production in BPD. Analysis of expression levels of key mitochondrial-related genes in the postmortem human brain has provided additional evidence to support this idea. A recent microarray study comparing postmortem hippocampal tissue from individuals with BPD and individuals with schizophrenia as well as normal healthy controls revealed that the expression of 43 genes was decreased in patients with BPD compared with those with schizophrenia (
49). Furthermore, 42% of these genes encoded mitochondrial proteins.
In addition to the prominent findings of altered expression of genes encoding mitochondrial proteins, substantial changes have been observed in the level of expression of proteins implicated in synaptic function. These findings suggest alterations in synaptic plasticity mechanisms in individuals with BPD. The neuronal plasticity marker GAP-43 is highly expressed in axonal growth cones during development and is implicated in regulation of axonal morphology and synaptic plasticity in the mature brain (
55). In patients with BPD, it has been reported that levels of GAP-43 are reduced in both the cingulate cortex and the hippocampus (
56,
57). The synapsin family of proteins binds synaptic vesicles to the cytoskeleton, preventing their transport to the presynaptic membrane and subsequent neurotransmitter release (
58). Docking of synaptic vesicles and neurotransmitter release are regulated by a complex of proteins that includes SNAP-25, syntaxin, synaptobrevin, and synaptophysin. In postmortem brains from individuals with BPD, a reduction in the levels of synapsin family members has been found in the hippocampus (
59). However, increases in the levels of SNARE complex proteins, which are responsible for mediating the fusion of synaptic vesicles with the cell membrane, have been observed in the DL-PFC (
60). An additional study showed decreased levels of synaptobrevin and synaptophysin in the visual association cortex of patients with BPD (
61). The levels of mRNAs encoding netrins, a family of proteins important in regulating axon guidance, have also been shown to be reduced in the CA3 region of the hippocampus and entorhinal cortex of patients with BPD (
62).
Genetic studies.
Combined evidence from family, twin, and adoption studies suggests that BPD has a strong genetic component. Twin studies show a highly elevated concordance rate in monozygotic twins when compared with dizygotic twins (
63,
64), and BPD is more likely to occur in biological parents of adopted children than in the adoptive parents (
65). Strategies for detecting the genetics of BPD include both linkage and association studies. Linkage methods test the loci of vulnerability genes by studying co-inheritance of chromosomal fragments in specific illnesses, whereas association studies test whether a specific gene variant is associated with a given disorder. A list of genes, including their known functions and potential role in the etiology of BPD, that have been implicated via both linkage and association studies is lengthy and beyond the scope of this review (see ref
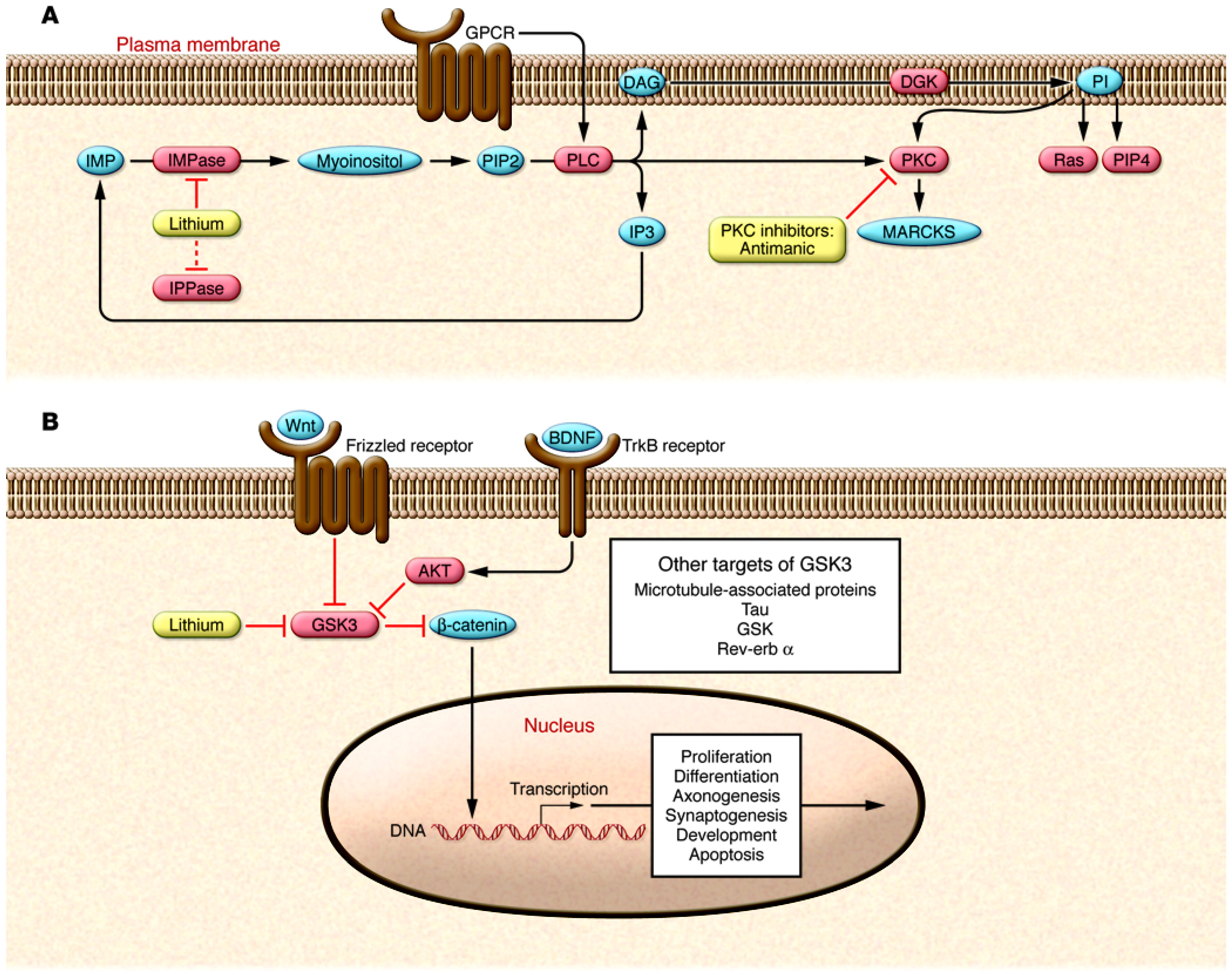
66 for an indepth discussion). Of the implicated genes, most attention has been given to those that encode proteins known to interact with signaling pathways previously implicated in BPD. For example, several of the risk genes that have been identified are known to interact with the PKC and glycogen synthase kinase 3β (GSK3β) signaling pathways (
67,
68). In addition, other putative susceptibility genes, including glutamate receptor, metabotropic 3 (
GRM3) and
GRM4; glutamate receptor, ionotropic,
N-methyl-D-aspartate 2B (
GRIN2B); D-amino acid oxidase (
DAO); and DAO activator (
DAOA, also known as
G72), encode proteins involved in glutamatergic signaling (
68,
69). In addition, a number of genes encoding proteins involved in circadian biology, including ARNT-like protein 1, brain and muscle (BmaL1), TIMELESS, and PERIOD3, have been implicated as susceptibility genes for BPD (
68). These genes are of interest because virtually all patients with BPD have alterations in circadian function, including alterations in sleep patterns, activity, hormonal secretions, and appetite.
Beyond the older linkage and association studies, recent advances in technology have allowed researchers to study the genetic component of BPD using genome-wide association studies (GWASs). Four groups have recently performed independent GWASs of BPD (
67,
70–
72). However, the significance of these findings is unclear, since very few findings have been replicated from sample to sample and there is the possibility of multiple false positives due to the substantial number of comparisons. In one study using 1,233 patients with BPD and 1,439 control subjects, Baum and colleagues identified a SNP in diacylglycerol kinase η (
DGKH) as being associated with BPD (
67). The Wellcome Trust Case Control Consortium (WTCC) analyzed 1,868 individuals with BPD and 2,938 control subjects and identified a locus in a gene rich region of high linkage disequilibrium on chromosome 16p12 as being associated with BPD (
70). Sklar et al.'s study of 1,461 patients with BPD and 2,008 control subjects found their strongest result in myosin 5B (
MYO5B) (
71). The most recent GWAS, by Ferreira et al., used a new sample with 1,098 individuals with BPD and 1,267 control subjects and found the strongest results in ankyrin G (
ANK3) and calcium channel, voltage-dependent, L-type, α 1C subunit (
CACNA1C) (
72). A broad comparison of the WTCC and the Sklar study also confirmed the
CACNA1C results, identifying
CACNA1C as showing a consistently strong signal in individuals with BPD (
72). More studies are clearly necessary before any consensus can be made as to the significance of these recent findings.