A notable source of morbidity and mortality, dementia accounts for an excess of health care costs.
1 The total economic cost of the disorder, including direct and indirect medical costs, has been estimated to be $100 billion per year (in U.S. dollars).
2 Alzheimer's disease (AD) is a progressive and incurable disease characterized by cognitive and behavioral abnormalities and is the most common cause of dementia in North America.
1 The estimated prevalence of AD among individuals above 65 years of age in the United States is 10.3%.
3The behavioral abnormalities associated with AD frequently disrupt patient care and present a management problem to the caregiver. Behavioral and psychological symptoms of dementia (BPSD) that complicate AD include behaviors with psychotic features (e.g., delusions and hallucinations) as well as those without (e.g., depression, anxiety, agitation, wandering, hostility, and uncooperativeness).
4 Disruptive agitated behaviors are common and occur in 40% to 90% of patients with AD at some point during the course of their illnesses.
5,6 Evidence suggests that BPSD have a detrimental impact on both patients
7 and caregivers.
8 Aggression occurs in 18% to 65% of patients
9–11 and is the behavioral disturbance that causes the greatest impact. Behaviors such as agitation and aggression complicate management
12,13 and contribute to caregiver burden
14 and the institutionalization of demented patients.
15–18The treatment of behavioral disorders in AD has emphasized the use of psychotropic medications.
19–21 Studies in nursing homes,
22–24 hospitals,
25 and the community
18 have shown that psychotropic medications are frequently used in elderly patients. Guidelines outlining the appropriate use of psychotropics in elderly institutionalized patients have been created in response to persistent psychotropic use.
26 The Omnibus Budget Reconciliation Act of 1987 was enacted to curtail inappropriate and overuse of psychoactive medications in long-term care facilities, which often resulted in high incidence of adverse events.
27,28 For example, antipsychotic medications, the most commonly used psychotropic for BPSD, can produce serious extrapyramidal side effects. Extrapyramidal symptoms such as rigidity, tremors, and parkinsonian gait
29are common with antipsychotic drug administration and may limit treatment in the elderly demented population and lead to discontinuation of medication. Psychotropics have also been found to induce agitation, confusion, and cognitive impairment in elderly demented patients,
30 further contributing to BPSD. Novel therapies targeting specific neurotransmitters are believed to underlie behavioral dysfunction in AD are being studied as alternatives to the modestly efficacious antipsychotics.
31,32 The idea here is that we can rationalize treatment by studying the neurotransmitters involved. This has been demonstrated with serotonin, one of the best examined neurotransmitters.
33,34METHOD
Neuropathological and neurochemical disruptions in the central noradrenergic system have been established as important factors in the etiology of AD and BPSD. Evidence supporting these neurobiological alterations as well as the putative links between the noradrenergic system and behavior, including BPSD, are reviewed. Noradrenergic pharmacotherapies are also examined to assess the clinical significance they may have in AD and BPSD. Sources used to obtain articles included electronic databases (MEDLINE, EMBASE) and manual cross-references from bibliographies of relevant literature. The following keywords were used: norepinephrine, noradrenergic, Alzheimer's disease, dementia, behavior, behavioral disorders, aggression, agitation, psychosis, depression, and noncognitive.
NOREPINEPHRINE SYSTEM
The central noradrenergic system is known to have two different projections: 1) neurons originating from the ventrolateral tegmental noradrenergic cells—involved in sexual and feeding behaviors
35—that flow to the forebrain, an area commonly involved in traumatic brain injury and associated with violent behavior and reduction in rage control,
36 and 2) neurons originating from the locus coeruleus (LC), which line the bottom of the fourth ventricle in the rostral pons
37 and are associated with cognitive functions.
38 Noradrenergic neurons give rise to diffuse axonal projections that innervate large areas of the brain, including the cerebellum, thalamus, hypothalamus, and midbrain,
35 and thus can be expected to influence many different functions.
Norepinephrine (NE) and epinephrine are catecholamines and are formed from the amino acid l-tyrosine which is actively transported from the blood. l-Tyrosine is converted to l-dihydroxyphenylacetic acid (l-dopa) through tyrosine hydroxylase, which is the rate limiting enzyme of the metabolic pathway. l-dihydroxyphenylacetic acid is then transformed to dopamine (DA), the immediate precursor of NE, by dopa-decarboxylase and transported into storage vesicles. Storage vesicles containing DA beta hydroxylase (DBH), found in NE-specific neurons, convert the stored DA to NE through β-hydroxylation. Dopamine producing neurons have storage vesicles without DBH.
Norepinephrine is the primary neurotransmitter of the sympathetic nervous system in the periphery but is also prevalent in the brain. Epinephrine is an analog of NE and is formed by N-methylation. It is released principally from the adrenal medulla; however, its function in the CNS has not been fully elucidated. Norepinephrine is released from the vesicles into the synaptic cleft at the occurrence of an action potential. Norepinephrine transporters on the synaptic terminals take up NE into storage vesicles. Monoamine oxidase (MAO) deaminates and, hence, deactivates free NE in the cytosol that has not been taken up into the vesicles, leading to the formation of the metabolite, 3,4-dihydroxyphenylglycol (DHPG). Dihydroxyphenylglycol is then converted to 3-methoxy-4-hydroxyphenylglycol (MHPG) by catechol O-methyltransferase (COMT). Residual NE in the synapse is first metabolized by COMT and then MAO to form MHPG and other metabolites.
Noradrenergic receptors are divided into α-adrenergic and β-adrenergic subtypes, each of which has further subtypes: α
1, α
2, β
1, β
2 and β
3. The postsynaptic β-adrenergic receptors activate G
s type G proteins to stimulate adenylyl cyclase, leading to a physiological response. The α
2 adrenergic receptors decrease adenylyl cyclase activity or activate specific K
+ channels through coupling to G
i and/or G
o,
39 and are located both pre- and postsynaptically. The α
1 adrenergic receptor stimulates the action of phospholipase C through G
x. and it is located on the postsynaptic terminals. Only α
2 and β
2 adrenoreceptors are located on both presynaptic and postsynaptic terminals. Norepinephrine and epinephrine stimulate both the α- and β-adrenergic receptors in the periphery and CNS with the effects, depending on the balance between the α- and β-receptors that are present and have been activated. Presynaptic α
2 adrenoreceptors modulate the negative feedback of NE release and are pharmacologically different from postsynaptic α
2 adrenergic receptors. Norepinephrine release can be enhanced by the stimulation of β
2 adrenoreceptors. Epinephrine is more potent than norepinephrine on the β
2 adrenoreceptor.
Norepinephrine mediates and is modulated by other neurochemicals in the brain, many known to affect behavior. A loss or alteration in any one neurotransmitter is likely to affect other neurotransmitters and potentially lead to deviations in behavior. The noradrenergic system interacts with acetylcholine,
40–43 serotonin,
44–46 DA
47–49 and γ-aminobutyric acid.
50 Other neurotrophic factors that interact with and modulate the noradrenergic system include a corticotropin releasing hormone,
51,
52 somatostatin,
53 and neuropeptide Y.
54 Many of these neurotrophic factors have been associated with behaviors such as aggression, anxiety, and depression in nondemented populations
55 and thus alterations in particular neuropeptides may play a role in aggravating already disrupted neurotransmitter systems. Since multiple neurochemical systems are known to be compromised in AD,
56 they may magnify behavioral disturbances.
NOREPINEPHRINE AND ALZHEIMER'S DISEASE
Alzheimer's disease is associated with multiple neurotransmitter system dysfunction. The most well studied neuronal system dysfunction is the cholinergic system. However, evidence supporting noradrenergic, serotonergic, dopaminergic, corticotropin-releasing factor, and somatostatin system dysfunction has also accumulated,
56–58 and mounting evidence supports the presence of significant abnormalities in the noradrenergic system in AD.
58–78Norepinephrine Concentrations
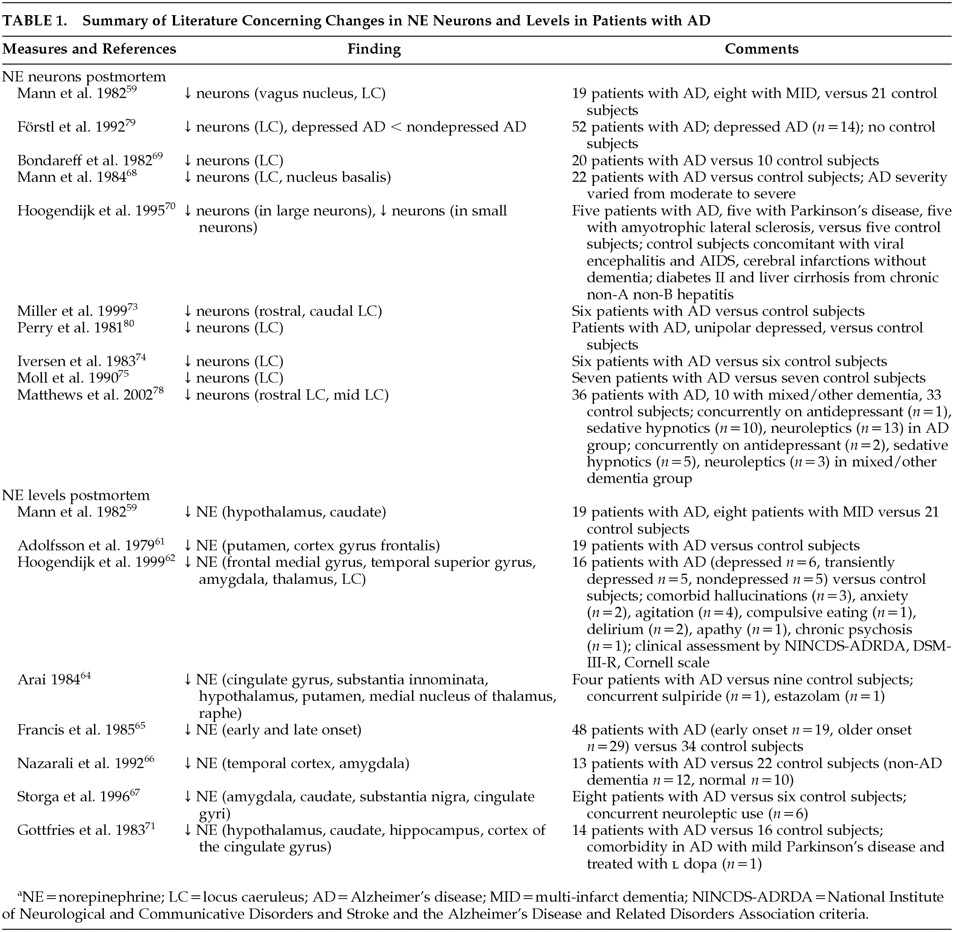
Postmortem studies have consistently shown noradrenergic system involvement in the AD disease process with decreased NE levels being recognized in many brain areas.
58,59,61,62,64–67,71,72,74–78 The majority of studies indicate significantly decreased cortical and subcortical NE levels in the frontal medial gyrus, temporal superior gyrus, cingulate gyrus, hippocampus, amygdala, thalamus, hypothalamus, caudate, putamen as well as the LC.
58,59,62,64,66,67,71,75,76 However, some studies did not replicate these results due to possible confounders such as postmortem delay.
Loss of Noradrenergic Neurons
A loss of noradrenergic neurons in the LC, the major noradrenergic source in the brain, has been well established in patients with AD,
59,68–70,73–75,79,80 and has not been found in other types of dementia such as vascular dementia.
59 These changes in the LC may account for a deficiency in the noradrenergic system in the pathogenesis of AD.
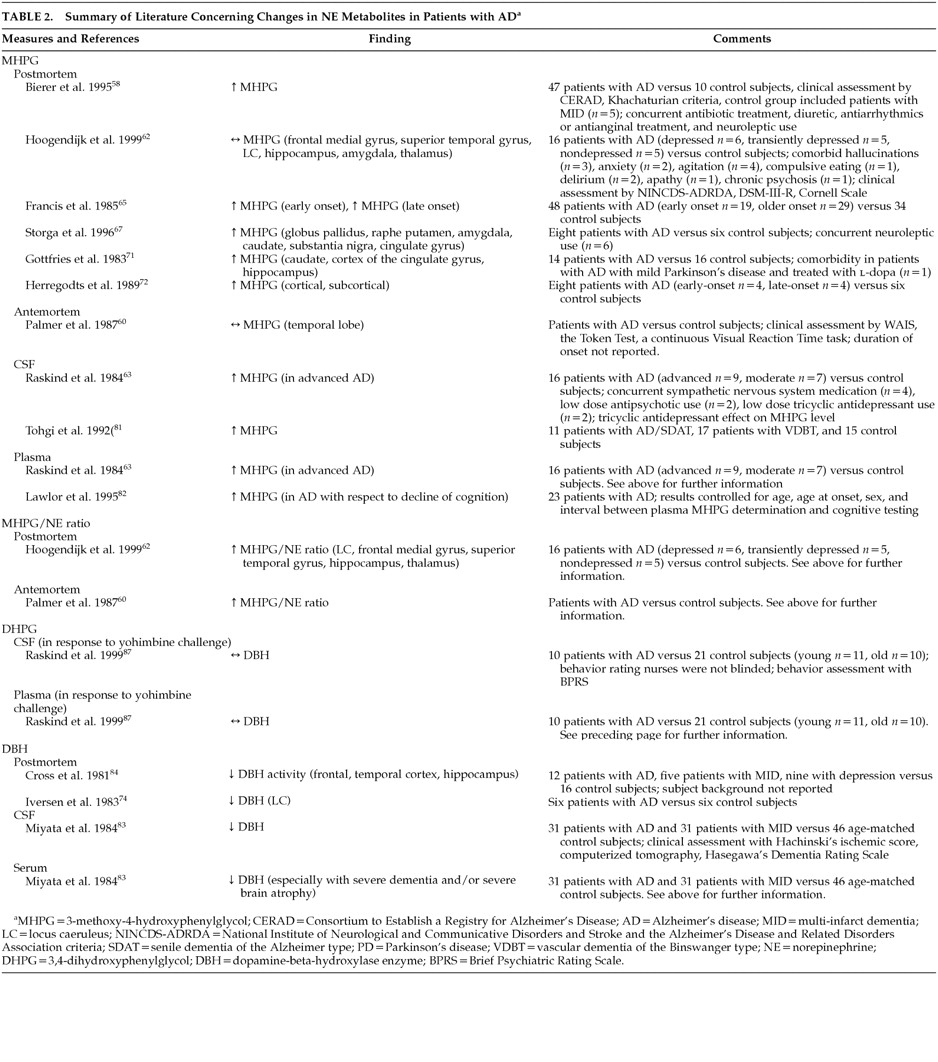
However, NE concentrations in tissue may not directly correlate with functional noradrenergic transmission. Rather, noradrenergic turnover and neuronal activity deduced through measurements of relative concentrations of NE and its predominant intraneuronal metabolite, MHPG, provide a more accurate representation of actual noradrenergic function. In postmortem studies, when both NE and MHPG were quantified together, although brain NE was often decreased, MHPG was found to be unchanged or high in AD patients compared to control subjects.
58,62,65,67,71,72,81 These findings suggest that neuronal loss may lead to subsequent increased NE metabolism and hence increased noradrenergic activity, possibly as a compensatory mechanism for LC NE loss. Hoogendijk et al.
62 demonstrated a significant inverse correlation between the MHPG/NE ratio and the number of remaining pigmented LC neurons in AD patients, inferring an overactivity or activation of the remaining LC neurons to counterbalance cerebral NE losses. It remains unclear whether this compensatory activity occurs in the noradrenergic neuron terminals in the projection areas, in the LC cell bodies or in both.
62Galanin (GAL), a major inhibitory modulator of cholinergic and noradrenergic transmission, has been shown to be hyperinnervated in the basal forebrain and cortex of AD patients. Galanin fibers are thought to originate in the LC. Miller et al.
73 explored the possibility that GAL expression is increased in the LC of patients with AD. Although the authors observed a significant reduction in the amount of neuromelanin-pigmented, which identifies NE presence, neurons in the LC of AD patients, when investigated alongside age- and sex-matched control subjects, there was no significant difference found between the number of both the pigmented and nonpigmented GAL mRNA expressing neurons in either demented patients or control subjects. This resulted in a significant increase in the percentage of neuromelanin-pigmented GAL co-expressing cells in patients with AD compared to control subjects, hence, suggesting preservation or sparing of noradrenergic neurons co-expressing GAL mRNA in a brain area otherwise diminished of noradrenergic neurons. These results indicate a possible neuroprotective feature of GAL, safeguarding the GAL co-localized group of noradrenergic neurons from damage resulting from the disease process.
73 The possibility that augmented GAL fiber innervation may be due to the enhanced expression of GAL within the remaining neurons in the LC region remains to be investigated further.
Link With Severity
There is a well-established link between AD severity and loss of noradrenergic neurons.
69 Bondareff et al.
69 compared total neuronal counts of 19 AD patients compared to 10 nondemented subjects and found the group of AD patients could be further divided into two distinct groups according to LC neuronal cell counts, which also correlated with severity of dementia. The authors reported an 81% decrease in nucleus LC neurons in the group of demented patients with more severe dementia, when compared to control subjects. The group of patients with less severe dementia showed a 20% decrease in LC neuronal loss. NE levels in the brain have also been found to have an inverse relationship with cognitive impairment.
61,78 Lawlor et al.
82 noted a significant positive relationship between basal plasma MHPG levels and cognitive impairment, further corroborating previous suppositions that increased MHPG from heightened NE turnover, as a result of losses of NE, is correlated with increased cognitive dysfunction.
Dopamine Beta Hydroxylase (DBH)
Studies of DBH may further substantiate evidence of noradrenergic dysfunction in AD. Both postmortem and antemortem studies have shown DBH, a noradrenergic biological marker, to be reduced in the hippocampus and frontal and temporal cortices of AD patients when compared to controls
74,83,84 and patients with vascular dementia.
83,84 However, these results must still be interpreted with caution as the extent to which DBH activity is associated with LC neuronal degeneration is still unclear.
84Cerebral Spinal Fluid (CSF)
Resting cerebral spinal fluid NE is thought to be generally increased with age, particularly in patients with advanced AD, and has been shown to be higher in AD patients when compared to control subjects.
63,85 This is consistent with the proposed compensatory action of noradrenergic neurons. It is essential to note that the major method of NE clearance from the CSF is through NE reuptake by presynaptic noradrenergic terminals.
86 Therefore, it is possible that AD related noradrenergic circuit degeneration may lead to impairment of NE reuptake resulting in increased CSF NE levels.
87 Hence, measuring CSF NE concentrations may not allow a direct evaluation of CNS noradrenergic dynamics. It is more important to measure NE precursors and metabolites as well CSF NE levels for a comprehensive analysis of NE activity.
88 Moreover, MHPG, unlike NE, crosses the blood-brain barrier freely, hence CSF MHPG can provide an indication of neuronal NE metabolism and dispersion of circulating MHPG into CSF. Therefore, it is imperative that plasma MHPG levels are accounted for in order to accurately interpret CNS noradrenergic activity.
63 Raskind et al.
87 compared levels of NE, its metabolite DHPG, and precursor dopa in both CSF and plasma following administration of yohimbine (a selective α
2 adrenergic receptor blocker), clonidine (a selective α
2 agonist), and placebo in older subjects with AD, older subjects without AD and younger subjects. The results suggested that the increase in CSF NE subsequent to yohimbine administration in both older subjects and AD subjects is not likely due to a reduction in NE clearance but rather mechanisms that compensate for NE neuron loss through augmented NE biosynthesis.
Cerebral spinal fluid NE response to manipulation of the α
2 adrenergic receptor by the administration of yohimbine and clonidine has been studied to evaluate noradrenergic tone, yet a very limited amount of evidence is available regarding noradrenergic dysfunction. A general increase in CSF NE has been seen in response to yohimbine in both AD and older control subjects (with a greater increase in the AD subjects), when compared to younger control subjects.
87,89 Results of studies of the CSF NE response to clonidine have been inconsistent, with insignificant results predominating. Further research into this area is needed to elucidate the value and capacity of CSF catechols as an index of neuronal neurotransmission function.
Compensating for lost neurons results in the tonic activation of the noradrenergic system leading to an increased noradrenergic response. Mechanisms that may account for this include the following: 1) Neuronal plasticity
38,89—resulting in the hyperinnervation of certain brain regions. This may be a result of regenerating neurons and has been observed in rats.
38,89 2) Increased NE synthetic capacity and firing rate in damaged LC neurons.
38,89 3) Decreased ability of presynaptic α
2 receptors to inhibit noradrenergic activity.
38,89 Studies have indicated a loss of α
2 receptor inhibition in AD, leading to the tonically activated state of the noradrenergic system and increased CSF NE levels.
87,89 This increase in compensatory activity is usually accompanied by compensatory activity in other neurotransmitter systems as well.
Both postmortem and antemortem evidence supports a consistent link between noradrenergic dysfunction and AD (
Table 1 and
Table 2). Postmortem studies provide the strongest evidence of noradrenergic degeneration in AD. Although postmortem evidence may provide a descriptive representation of the loss of noradrenergic neurons, postmortem results may not directly reflect the antemortem state. Nonetheless, much of the evidence reviewed reveals decreased levels of both NE and noradrenergic neurons, while MHPG levels have been found to be increased, suggesting increased NE metabolism and, hence, activity. The fact that this correlates positively with increased AD severity, further validates these findings.
NOREPINEPHRINE AND BEHAVIOR
The LC is sensitive to both variations in the body's internal homeostasis and external environmental stimuli, which can activate not only the central noradrenergic system and sympathoadrenal system but may result in behavioral manifestations as well. The LC is involved in regulation of levels of arousal, flight-and-fight responses,
89,90 agitation, anxiety,
89 sleep-wake cycle, and levels of vigilance and emotion
70,91,92 as well as aggressive behaviors.
93–95 Noradrenergic projections from the LC also modulate sympathetic nervous system response, including blood pressure, pulse rate and danger signals. Hence, the noradrenergic system, directly or indirectly, has the potential to modify a variety of behaviors through actions in both the cortical and subcortical regions. A disruption of such an integrated system could potentially lead to abnormal behavioral responses to otherwise ordinary stimuli. LC damage has been determined in several neurologic disorders that are characterized by altered behavior including Parkinson's disease, schizophrenia and depression.
70,92The link between NE disruption and behavior has been studied in a variety of psychiatric disturbances including depression, anxiety, agitation and aggression. In depression there is evidence of α
2 receptor dysfunction including postsynaptic α
2 receptor downregulation,
96–98 increased presynaptic α
2 receptor sensitivity,
99 and increased α
2 receptor density in the LC.
100,101 Decreased noradrenergic receptor sensitivity and increased noradrenergic turnover have been noted in patients with anxiety,
102 generalized anxiety disorder,
103 and posttraumatic stress disorder.
104 High levels of NE release have been associated with aggression in animals, healthy adults
93,105,106 and patients with depression and mania.
107,108 In animal models, NE enhances aggressive behaviors
106,109 and β-adrenoreceptor blockers have been shown to decrease aggression.
106 Finally NE and NE metabolite levels are positively correlated with aggressive behaviors in patients with personality disorders.
110–112 In summary, there is significant evidence of noradrenergic system involvement in a variety of psychiatric and behavioral disorders from animals and nondemented human studies.
NOREPINEPHRINE AND BEHAVIOR IN DEMENTIA (BPSD)
As previously mentioned, behavioral problems, particularly agitation and aggression are prevalent in AD. This has been consistently shown in various studies.
113–116 A prospective, 10-year, longitudinal study on the change in aggressive behavior throughout the course of dementia by Keene et al.
117 measured physical aggression, aggressive resistance, physical threats, verbal aggression, refusing to speak, destructive behavior and general irritability. The authors reported 96% of the 99 subjects with AD/vascular dementia showed profound or continuing aggressive behavior of at least one type during the dementia disease process, with verbal aggression as the most common behavioral disturbance. As the noradrenergic system has been implicated in the disorder of behavior in animals as well as other psychiatric ailments, it has been suggested that NE may likely play a role in the noncognitive behavioral disturbances associated with AD.
118 Agitation, particularly aggression and akathisia, has been linked with increased norepinephrine activity.
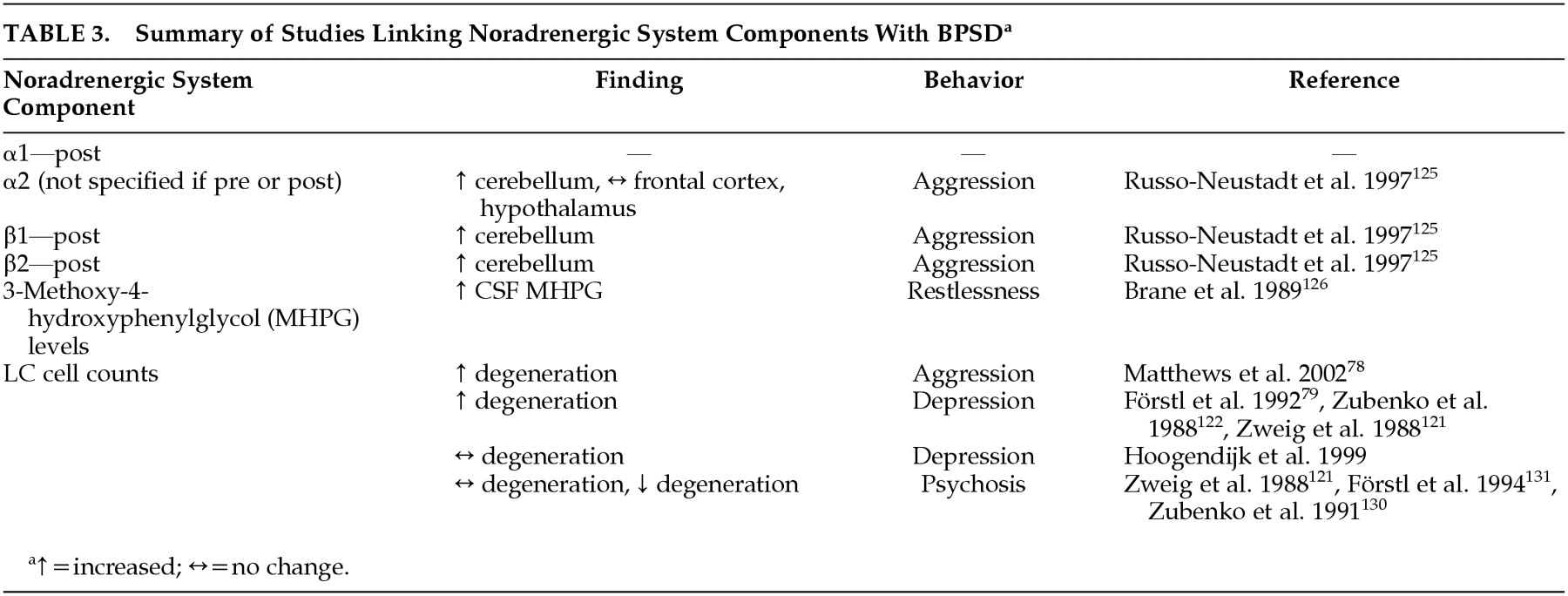
119 Noradrenergic system components have been associated with behaviors associated with dementia (
Table 3). The role of norepinephrine in the facilitation of BPSD in AD is reviewed below.
Depression
Depression may complicate AD and has been found to be most common among people with mild severity of dementia.
120 Both AD and depression share similar signs and symptoms, including loss of interest and appetite, apathy, and sleep disturbances.
4 Studies have found that AD patients suffering from comorbid depression have significantly more LC neuron degeneration when compared to nondepressed AD patients.
79,121,122 However, contradictory evidence exists. Hoogendijk et al.
123 also compared LC neuronal cell counts between depressed and nondepressed AD patients but found no significant differences in LC degeneration between depressed and nondepressed AD patients. This may be due to methodological differences between the studies such as confounding by level of severity of both dementia
121 and depression,
121,122 LC sample section size and location of analysis.
79,122 A reduction in NE has also been observed in cortex of depressed AD patients
124 further supporting the role altered noradrenergic function may play in influencing depressive symptoms. Thus, the literature currently supports an association between NE and depression in AD.
Aggression and Agitation
Russo-Neustadt and Cotman
125 observed a small but significant increase in total β-adrenoreceptor concentration in the cerebella of aggressive AD patients when compared to nonaggressive AD patients and healthy control subjects. This finding suggests a possible link between aggression/agitation and β-adrenergic receptor binding.
Since presynaptic α
2 receptors regulate the negative feedback of NE and postsynaptic receptors are thought to regulate postsynaptic neuronal function, the differential location of the α
2 adrenoreceptor may play a role in modulating aggression.
106 Activation of the α
2 adrenoreceptor by an agonist may lead to differing results depending on whether the post- or presynaptic receptors were stimulated or, possibly, depending on the ratio of the activated receptors. A postmortem study
125 of the frontal cortex, hypothalamus and cerebellum (areas innervated by LC neurons) of agitated AD patients, nonagitated AD subjects and healthy elderly control subjects revealed a significantly elevated level of α
2 receptors in the cerebellum of aggressive subjects, 70% higher than the nonagitated AD subjects. The levels of α
2 receptors found in the nonagitated AD patients were slightly but not significantly higher than the levels found in the healthy elderly control subjects. Measurements of the α
2 receptors in the frontal cortex and hypothalamus were not found to vary significantly.
There is some evidence that supports an association between motor restlessness and increased MHPG levels. MHPG levels have been found to be correlated with restlessness in AD patients.
126In vivo studies have also been performed to address the link between BPSD and NE. Yohimbine has been reported to lead to increased release of NE and successive activation of the postsynaptic α
1 adrenoreceptors.
127 It has been shown that AD patients are more sensitive to administration of yohimbine, compared to other elderly patients and younger subjects,
87,128 and exhibit more agitated behaviors. This may be due to increased postsynaptic noradrenergic sensitivity in AD.
87,89 This is plausible, as postsynaptic β-receptors have been reported to be up-regulated in the prefrontal cortex and hippocampus of AD patients,
89 consistent with the postulated increase in sensitivity to noradrenergic stimulation.
87 As a result of noradrenergic overactivation, AD patients would no longer be able to focus attention and their mechanisms of coping with stressful stimuli would be compromised. Even in the absence of stressful stimuli, the noradrenergic system would be active. This may account for the aggression displayed in many AD patients.
38,62 These findings further consolidate the probability of an adrenergic dysfunction contributing to the pathophysiology of agitated behaviors in AD.
Psychosis
Psychosis is a cluster of clinically disruptive behaviors often observed in AD patients. Psychotic behaviors involve delusions and hallucinations and can aggravate aggression.
129 Both evidence supporting the association between psychotic behaviors and noradrenergic preservation,
130 and evidence showing no association between psychosis and NE
121,131 have been published. Zubenko and associates
130 found higher NE concentrations in the substantia nigra of psychotic AD patients when compared to nonpsychotic AD patients. Förstl et al.
131 found that AD patients with auditory hallucinations or delusions had significantly higher neuron counts in the parahippocampal gyrus and a trend towards lower dorsal raphe nucleus neuron counts compared to AD patients without psychosis; however, this was not observed in the LC. The link between the noradrenergic system and psychotic behaviors is still unclear and further investigation is necessary.
In summary, evidence from both animal and human studies supports a link between noradrenergic dysfunction and behavior, particularly aggressive behavior. Depressed patients, patients suffering from anxiety disorders and AD patients all show blunted growth hormone (GH) responses to clonidine, strongly suggesting a disruption in noradrenergic control. Intensified noradrenergic activity and/or hypersensitive adrenoreceptors are thought to mediate this NE dysfunction in aggression associated both with and without AD as well as anxiety and depression. It is believed that compensation for the loss of the noradrenergic neurons that occurs during the progression of AD leads to an increase in the number of noradrenergic receptors and/or increased activity of remaining noradrenergic neurons, the potential source of aggressive behavior. This is credible as β1, β2 and α2 receptor upregulation has been shown in aggressive AD patients when compared to nonaggressive AD patients. The clinical expression of aggression likely is also dependent on other neurotransmitters. In summary, it has been shown that both α and β-adrenoreceptors are involved in the modulation of aggression. However, the exact role each receptor plays is still uncertain and further research is needed to clearly assess the link with NE and behavior, particularly BPSD.
NORADRENERGIC PHARMACOTHERAPY
The probability that the noradrenergic system contributes to the pathophysiology of BPSD associated with AD has led to potential therapeutic interventions. Pharmacological manipulation of the CNS noradrenergic system through the blockade of the beta adrenergic receptors has been studied as a treatment for a variety of neuropsychiatric disorders.
132 As it is believed norepinephrine is involved in the modulation of behavior, noradrenergic agents have much pharmacological potential for the amelioration of behavioral disturbances. Studying the effects of noradrenergic agents on the various behavioral disturbances associated with dementia can also lead to a better understanding of their neurochemical etiology in the way neuroleptic response led to an understanding of the role of DA in schizophrenia.
Beta Blockers
Previous studies have indicated propanolol, a long-acting β-blocker, to be an effective treatment for aggression and agitation in patients with dementia who have been unsuccessfully treated with conventional therapies.
133–137 In a randomized double-blind crossover placebo-controlled study, Greendyke et al.
133 evaluated the efficacy of propanolol for an 11-week period at a maintenance dosage of 520 mg/day, in assaultive patients with organic brain impairment. The authors reported significantly fewer assaults and attempted assaults by the nine patients who completed the study, during the active treatment phase when compared to the placebo phase. Five patients showed marked improvement, two showed moderate improvement and two showed little or no improvement of violent behavior. A similar study was repeated by Greendyke and Kanter
138 using pindolol, another β
1 blocking agent, rather than propanolol. Pindolol wields a partial agonist effect thereby exerting similar therapeutic benefits as propanolol but with less cardiovascular side effects such as hypotension, bradycardia and decreased cardiac output. Patients were maintained on an individualized optimum dosage between 40–60 mg/day for a minimum of 10 days. Significant reductions in assaultiveness, hostility, lack of communication, uncooperativeness and repetitive behaviors were noted. Shankle et al.
135 observed a 71% response rate in patients with either AD or vascular dementia (VaD) with both agitated and aggressive behaviors who were treated with propanolol. The 12 patients received 20 mg propanolol per day which was increased to an efficacious maximum that ranged between 30 and 80 mg per day. One patient suffered an adverse event (bradycardia), which was resolved by lowering the dose. A case series by Weiler et al.
137 also demonstrated success in relieving treatment refractory disruptive behavior in 4 patients with AD and 2 patients with VaD. The propanolol doses ranged from 80 mg/day to 520 mg/day and no adverse events were noted. These studies indicate that the modulation of the β-adrenoreceptors through the use of β-blockers has been useful in the reduction of aggressive behaviors. However, many of these studies involved patients with varying dementia etiologies. A randomized controlled trial with AD patients treated for BPSD using β-blockers is necessary before one can accurately judge effectiveness. Important clinical considerations must be recognized when using β-blockers in elderly patients. β-Blockers may exacerbate various concomitant illnesses such as chronic obstructive pulmonary disease, diabetes mellitus and peripheral vascular disease. Additionally, beta-blockers have been shown to increase plasma concentrations of some antipsychotics, including chlorpromazine and thioridazine.
139Antidepressants
Imipramine, a tricyclic antidepressant, inhibits the reuptake of noradrenaline and has been shown to prevent upregulation of β-adrenoreceptors induced by stress
140 and may hinder the induction of tyrosine hydroxylase in the LC following cold stress,
51 suggesting a lessening of stress-induced norepinephrine release. Reifler et al.
141 compared the use of imipramine with placebo in 61 AD patients, of those, 28 had a diagnosis of depression and 33 did not. The authors found that the depressed patients scored significantly higher than the nondepressed patients on the Hamilton Depression Rating Scale and though all patients improved significantly during the study, the depressed patients improved significantly more than the patients without depression. Although clinical recommendations have been made about avoiding the use of tertiary amines in the elderly demented population,
142 imipramine was found to be extremely well tolerated in Reifler's study, as no significant differences in adverse events were noted in the imipramine-treated group compared to the placebo-treated group.
Mirtazapine is a noradrenergic and specific serotonergic antidepressant that directly blocks the pre- and postsynaptic α
2 receptors and antagonizes the 5-hydroxytryptamine (5-HT)
2 and 5-HT
3 receptors with high affinity and 5-HT
1 receptors with low affinity.
143 Mirtazapine has been used with favorable results in a case series by Raji and Brady
144 in AD patients suffering from depression as well as anxiety, weight loss and insomnia. Three patients were given a trial of mirtazapine. Two patients were started at 7.5 mg and titrated up over 2 weeks to 15 mg. One of those patients was further titrated up over 2 more weeks to 30 mg. The third patient was started at 15 mg and was increased to 30 mg after 2 weeks. No changes were noted in cognition; however, improvement in appetite and sleep was documented within 2–4 weeks and cessation of anxiety and depression was noted at 2 months. None of the patients suffered any adverse effects. This case series suggests that mirtazapine may be an effective therapy for depressed AD patients with comorbid insomnia, anxiety and appetite loss. However, there is a lack of randomized controlled trials using mirtazapine, and therefore the extent of the usefulness of mirtazapine pharmacotherapy is still unknown and remains to be explored.
Other antidepressants with putative effects on the NE system include venlafaxine, bupropion, and reboxetine, though none of these agents have been tested for BPSD.
Psychostimulants
Psychostimulants, such as methylphenidate (MPD), have been used in the past to treat affective disorders, particularly depression. MPD is believed to block both norepinephrine and DA uptake and promote catecholamine release.
145 Few controlled studies have evaluated the efficacy of psychostimulants in the treatment BPSD.
A case series by Maletta and Winegarden
145 examined three severely demented patients, suffering from apathy and anorexia resulting in weight loss, using methylphenidate therapy. Two patients were started on 5 mg bid and one patient was started on 5 mg once daily. Doses were increased up to the maintenance dose of 10 mg bid. Appetite was found to increase within 3–7 days of initiating therapy, and complete resolution of anorexia, leading to weight gain, was observed within a few weeks. Improvement in social interaction and hence a decrease in apathy was also noted by the nurses. The patients were maintained on methylphenidate for 9, 16 and 24 months, respectively, and were eventually tapered off successfully. No adverse effects were noted and no other psychotropic was used during the period of treatment. Kaufmann et al.
146 also found favorable results with one dementia patient who was treated successfully for depressive symptoms including appetite loss and apathy, using methylphenidate.
Branconnier and Cole
147 also conducted a placebo-controlled study in demented, apathetic patients using 20–30 mg MPD and found an improvement in apathy that correlated with a lessening of depressive symptoms. Galynker et al.
148 treated 12 AD patients and 15 VaD patients using methylphenidate and found a significant improvement in negative symptoms.
Although the benefits of methylphenidate therapy in the elderly have been reported for the last 50 years, the results are limited by poor designs and lack of standardized assessments for behavior and even diagnoses. Methylphenidate appears to be useful in alleviating depressive symptoms including appetite loss and apathy; however, more research is needed to explore the value of; MPD in the pharmacotherapy of other behavioral disturbances associated with AD. Furthermore, as noted previously, psychostimulants have effects on other neurotransmitters, and it is not clear whether their beneficial effects for BPSD are due to their actions on NE or other systems.
It is difficult to treat BPSD pharmacologically as some patients may be very sensitive to first-line therapy with antipsychotic medications. Treatment emergent side effects may be intolerable resulting in the discontinuation of the drug. Hence, prescribing medication for BPSD often involves trying a series of medications on each patient. The use of β adrenergic antagonists for BPSD such as agitation and aggression may be clinically advantageous due to the absence of the extrapyramidal symptoms and sedation commonly seen with antipsychotics. Other adrenergic agents have been studied for the alleviation of other behavioral symptoms frequently associated with dementia, however, the vast majority of noradrenergic agents prescribed are for depression. Although the pharmacological management of depression with noradrenergic drugs has been shown to be successful, there is a great need for further research to elucidate the role of noradrenergic pharmacotherapy in the treatment of depression associated with AD. Given the modest efficacy of antipsychotics and the variable responses to antidepressants, randomized controlled trials of noradrenergic agents for BPSD are urgently needed.
CONCLUSIONS
The bulk of neuropathological studies in AD show noradrenergic neuron degeneration, decreased NE levels and increased NE metabolites. This evidence strongly and consistently suggests enhanced NE turnover. The vast majority of studies; however, have been done postmortem, and consequently caution is necessary in their interpretation. As altered behavior may be a result of “state” rather than “trait” phenomenon, it is important that the behavior being examined is manifested at the time of death, so as to accurately measure the neurological changes believed to be responsible. Postmortem delay is a confounding variable in neurophysiological analysis as postmortem results may not correctly represent the antemortem state. The consequences of delay in freezing brain tissue after death and prolonged preservation of brain tissue prior to catecholamine analysis have not yet been fully accounted for and may limit the usefulness of postmortem studies.
Norepinephrine is involved in the sympathetic “flight-or-fight” response and thus is sensitive to environmental challenges and can modulate behavior accordingly. The noradrenergic system has been shown to mediate behavior, particularly aggression, in animals as well as in psychiatric illnesses. Although there is substantial evidence correlating NE with aggression, a direct link between NE function and aggression associated with dementia needs to be examined further. More research into identifying the role of the noradrenergic receptors and their subtypes in controlling behavior in AD is necessary to elucidate the extent behavior dysregulation may be accounted for by noradrenergic dysfunction. A limitation of the current knowledge of the role of NE in BPSD is lack of information on other behaviors such as apathy, wandering, and sexually disinhibited behaviors. The current literature search revealed studies that addressed depression, agitation, aggression and psychosis only.
Drugs that target NE have been successfully used in treatment of depression, which is commonly found in AD as well. However, there is little research, especially randomized controlled trials, into the use of noradrenergic treatments for BPSD. Studies on beta blocker treatment for aggression and psychostimulant use for affective disorders appear to be the better studied pharmacologic interventions in AD; however, they too suffer from lack of standardization in behavioral assessments, heterogeneity in dementia diagnoses and small sample sizes. Further comprehensive research is necessary to explore the potential clinical utility of noradrenergic agents for the pharmacological management of disruptive behaviors associated with dementia.
The study of the neurobiology of BPSD in AD is fraught with methodological difficulties. While much has been learned over the past two decades, the goal of a unitary etiological hypothesis may never be reached given the heterogeneity of AD and its variable effects on behavior. Providing putative links between discrete neurotransmitter dysfunction and behavior; however, will allow for more targeted pharmacological interventions. Only then will treatment of BPSD be truly “rational.”
ACKNOWLEDGMENTS
This study was funded in part by a grant from the Physicians' Services Incorporated Foundation, grant number 98-39.
The authors thank Luis Lewis, Jr. and Florance Chan for their assistance.