The randomized, double-blind, placebo-controlled trial is the gold standard for estimating drug treatment effects. This design may not be optimal for conditions in which a majority of patients will not participate because of concerns in stopping other medications or because of other clinical conditions prohibited by the protocol. An example of this occurs in Tourette’s syndrome (TS), a condition in which multiple concurrent brain disorders may be treated medically as part of routine clinical practice.
1 Other examples include adult and childhood epilepsies,
2–4 multiple sclerosis,
5,6 cerebrovascular diseases,
7–9 Parkinson’s disease,
10,11 and Huntington’s disease.
12 This article reviews current difficulties with randomized, double-blind, placebo-controlled trials in TS and presents a framework for considering specific conditions under which an alternative study design—a randomized, rater-blinded, active comparator design—may be useful. The objective is to improve our ability to estimate medication effects in a broadly representative sample of subjects in more general clinical conditions. Potential scientific, ethical, economic, and safety issues are also discussed.
TS
Tourette’s Syndrome is a neuropsychiatric disorder characterized by more than 1 year of motor and vocal tics that are functionally or socially disturbing. Prevalence estimates vary widely, from 0.1% to 3.8%.
13,14 Approximately 85% of patients who present for medical attention for TS also have attention deficit hyperactivity disorder (ADHD), obsessive compulsive disorder (OCD), or other psychiatric disorders.
15 Tics are commonly benign, but in more severe cases they can be socially impairing, functionally impairing, or painful, such that functioning at school or work is difficult or impossible. In these settings, tic suppression is a reasonable therapeutic goal.
Short- and long-term effectiveness of current medical treatment practices in TS are based on data primarily from studies involving 60 patients or fewer. These studies may report on changes in multiple symptom domains, but they mainly target single symptoms and do not allow treatment of comorbid diagnoses. Robertson
1 recently published a review of data from 24 peer-reviewed, short-term, clinical trials of tic suppression over the past 20 years involving 17 agents in 12 classes (e.g., stimulants, alpha-2 agonists, neuroleptics, atypical neuroleptics, selective serotonin reuptake inhibitors), on a total of fewer than 600 subjects. Thus, there are a variety of treatment options available for consideration. Regarding clinical decision making, however, there is, unfortunately, very little published data comparing treatment choices for tics, ADHD, or OCD in TS. There is even less data available to guide combination therapy for multiple diagnoses. Consequently, decisions clinicians make daily between mild or potent tic suppressors, ADHD treatments, or anti-OCD medications are based on accumulated anecdotal experience, folklore passed along by mentors in training, or industry sponsored marketing materials.
Limitations of Placebo-Controlled, Monotherapy Clinical Trial Designs in TS
Typically, clinical trials, particularly premarketing studies, are used to compare symptoms on a single study medication versus placebo. In addition, the requirement that subjects discontinue all other neuropsychiatric medications allows for a statistically valid assessment of the effect of drug on symptom severity and careful assessment of single drug adverse events, but it does not accurately reflect routine clinical practice. Because the majority of TS patients have multiple, treatable symptoms, clinical practice frequently involves concomitant use of multiple medications.
The standard practice in most rigorous clinical trials in TS using placebo-controls is monotherapy, without concurrent psychoactive medication. This practice creates important limitations that reduce the utility for clinical decision making in routine medical practice. Such limitations include:
1. The requirement that concurrent medications be discontinued means that many subjects with significant comorbid disease or severe tics will choose not to participate. This introduces selection bias that decreases the generalizability of a trial’s results into clinical practice.
2. At the conclusion of a positive trial, clinicians who wish to try the medication on patients taking multiple medications have no information on harmful or beneficial drug interactions.
3. The efficacy of the drug used alone may be greater than the efficacy of the placebo among patients willing to participate, but this does not prove that the effectiveness of the drug in a real-world population, including many concurrently medicated patients, is better than the effectiveness of the placebo. Conversely, a drug which appears to be no better than placebo when used alone in a selected sample in a randomized clinical trial may be beneficial when used in combination with other widely used medications in a more representative sample of patients.
Placebo-controlled, monotherapy trials in TS also create important ethical issues, some of which pose difficult choices for families and clinical researchers:
1. For those families who do choose to participate, weaning off current medications can be associated with significant exacerbation of ADHD, OCD, other behavioral symptoms, and tics.
2. Symptoms of comorbid disorders that are not targeted by the trial may worsen during the pretrial washout, which is a period of weaning off other medications, and continue to be poor during the drug or placebo treatment phase.
3. Families of subjects who would be harmed by discontinuing medications for concomitant disorders do not have access to the opportunity to participate in a trial.
As a result of these and other difficulties, large studies in TS are rarely performed, and those that are performed tend not to enroll patients with severe tic symptoms or multiple comorbidities (i.e., the patients who most need effective treatment). Placebo-controlled studies enroll just a small fraction of available patients. For example, the Treatment of ADHD in Children with Tics (TACT) clinical trial, which attempted to treat tics as well as comorbid ADHD and included a combination therapy treatment arm, required 12 large TS clinics a period of 39 months to enroll only 136 patients.
16In addition, further evidence to guide treatment from large (>100 subjects), double-blind, randomized, controlled clinical trials in TS is not likely forthcoming, for several reasons:
1. The payoff for pharmaceutical companies to conduct a premarketing study to acquire an indication in TS is low, given the low prevalence of tics-only TS that is severe enough to treat. To our knowledge, the Orphan Drug Act, designed to encourage trials for low prevalence disorders, has only been used to test one agent, pergolide, for TS.
17 Only one pharmaceutical company has conducted a large (N=148) United States premarketing study in TS and tic disorders. The results have been presented in abstract form
18 but have not yet been published.
2. The utility for pharmaceutical companies to perform active comparator studies between patented versus off-patent medications is probably low under most circumstances because there is no assurance of achieving superiority or even equivalence.
3. With the exception of the TACT study, the National Institutes of Health (NIH) have funded no large multicenter treatment trials in TS.
4. The motivation for families to participate in postmarketing studies that compare an approved drug (for another indication) to placebo may be low since the families could easily obtain a prescription for the medication without participating in a study.
The result of many of these difficulties is that investigators have turned often to alternative designs with lesser scientific value, such as quasi-experimental, observational, or retrospective studies and open-label studies of single agents. While such studies are common in TS,
19–29 their outcomes cannot be proven to be attributed to treatment interventions. Since TS severity fluctuates and patients may be more willing to participate during exacerbations, spurious positive results may occur because subsequent remission may represent regression toward the mean rather than treatment benefit.
Rationale for Alternative Clinical Trial Designs in TS
The paucity of large controlled clinical trials in TS that provide useful evidence for medical decision making in practice suggests that alternative study designs may be preferable in providing clinically useful evidence for treating TS. Ideally, to understand optimal clinical practice in TS and increase enrollment and generalizability of results, more TS studies should allow for concomitant treatment of comorbid disorders, reflecting routine clinical practice. In order to accomplish this efficiently and economically, an alternative study design that diverges as little as possible from routine clinical care and yet occurs within the real-world setting of the clinic may be needed. Frequent, intensive, and expensive study visits that are required to assess safety and efficacy in premarketing, randomized controlled trials are important for some, but not all, clinical trials.
There are two additional important rationales for alternative study designs. First, as advances in neuroscience increase the number and cost of treatments for neurological and psychiatric disorders, the importance of the need to assess quantitatively and efficiently the long-term benefits also increases. Second, NIH has embarked upon a much publicized “Roadmap” to enhance the efficiency of clinical research and increase the numbers of patients participating in clinical trials (http://nihroadmap.nih.gov/). This plan includes promoting clinical research networks “capable of rapidly conducting high-quality clinical studies and trials where multiple research questions can be addressed.” Increasing participation rates among patients and families is essential to achieve these objectives. Designing short- and long-term studies with high acceptability to patients and families and high participation rates will increase the likelihood of achieving these goals.
Proposal for Implementing a Randomized, Rater-Blinded, Open-Label, Active Comparator Trial in TS and Other Conditions With Multiple Neuropsychiatric Diagnoses
Due to the difficulties with placebo-controlled, monotherapy trials described in the prior sections, we propose that active comparator trials that allow for concurrent treatment of comorbid disorders may increase the economy and generalizability of clinical trials in selected circumstances. The challenge is to maintain scientific rigor while reducing the many inherent difficulties.
The benefits of a double-blind study design using medication administered through a study pharmacy include reduction in biased reporting of benefits and side effects. Such a design is compatible with the active comparator design and with allowing for concurrent treatments. Thus, such a design is consistent with our goals for improving TS research. We recommend, however, that two modifications of this approach may increase the economy or feasibility of clinical TS research. The first modification would be to administer medication through the patient’s pharmacy as part of routine clinical care. The second modification would be to employ a separate blinded-rater, leaving the prescribing physician and patient unblinded. For the remainder of this discussion, “rater-blind” will refer to a study design in which the physician and patient are aware of the active treatment allocation, but the study person assessing treatment response and side effects is not: that is, the rater is blinded.
Administering study medication through routine prescribing and employing an independent, blinded rater may increase the ability of individual or small groups of physicians with limited funding to perform clinical research. In practice, the main economy of the blinded-rater design relates to 1) elimination of the use of the study pharmacy to prepare and dispense drugs along with associated paperwork and fees; and 2) elimination of medication costs. In the case of generic drugs such as haloperidol ($8.99 for 90 2-mg tabs), this cost can be pennies per pill, but in the case of newly marketed, brand name drugs such as risperidone ($429.01 for 90 2-mg tabs) or ziprasidone ($243.99 for 60 20-mg tabs), this cost is not trivial (prices listed on www.drugstore.com website at time of writing). In addition, successful integration of this design into routine clinical care reduces the need for study-funding to support the clinician researcher’s salary for patient assessment time.
Reduction in costs might enable research in more cases to be performed by individual investigators or small groups of investigators without NIH grant funding. Many clinically important questions that lack major impact on public health could be addressed more easily with less expense. Because of the many possible threats to validity that result from removal of blinding of the investigator or patient, we will address practical aspects of this design in some detail.
Assessment of treatment outcomes using a blinded rater has been performed in randomized clinical trials for neurological and psychiatric diseases.
30,31 Although blinded raters have been used in assessments of videotapes in TS,
32 no large, randomized, rater-blind, active comparator trials have been performed in TS.
Proposed Sequence of Events for Randomized, Rater-Blind, Active Comparator Trials in TS
1. Educational information is given to patients during routine clinic visits about the need for and value of clinical research.
2. After clinical trials receive Institutional Review Board (IRB) approval, all patients who might become eligible during the study period (i.e., even those with mild current symptoms and/or adequate treatment) are approached, and informed consent is performed for all subjects willing to be enrolled in a study for prospective data collection on symptom severity.
3. Data on symptom severity are collected at regular clinic visits using standard brief questionnaires. The patient is evaluated and followed per routine clinical practice to provide baseline data.
4. Once a target symptom (e.g., tics) develops or worsens so that a treatment is indicated in accordance with routine clinical practice (e.g., tics worsen and cause physical discomfort daily), the appropriateness of the target symptom for treatment is reviewed by the physician and patient/family as part of standard medical care.
5. The details of the study are reviewed with the patient, including the existence of equipoise between available treatment options, and informed consent is obtained for the relevant treatment study.
6. The study compares two treatments, A and B, to which the following generally apply:
One or both are treatments that are already-marketed and in routine clinical use for the commonly occurring target symptom in TS, or one or both treatments are marketed for other indications, but a cohesive scientific theory or common clinical practice supports their use for a target symptom in TS.
With regard to efficacy and side effects in a representative sample of TS patients, including patients with comorbid diagnoses and patients taking multiple medications, there is equipoise, and the treating physician agrees that there is equipoise.
One or both treatments are unlikely to be the subject of industry-funded trials.
7. Subjects are randomized to active treatment A or B for the target symptom.
8. Treating physicians are aware of treatment assignments after randomization, as in routine clinical practice, and monitor for efficacy and side effects, as in routine clinical practice.
9. Medications are prescribed as in routine clinical use and paid for by the patient’s insurance company or other payer. Doses may be changed if clinically indicated.
10. Patients will continue concomitant medications for other symptoms. Use of other medications by name and class (e.g., fluoxetine, SSRI) is coded in the study record.
11. Brief, easily administered scales for symptom severity of the target symptom and commonly co-occurring symptoms and side effects will be used as outcome measures. For example, in TS studies, analysis should, at a minimum, assess tic, ADHD, and OCD symptoms with accepted rating scales.
12. Research outcomes are assessed by an independent rater, trained in administration of the clinical scales, blinded to treatment assignment and compliance. Baseline ratings are performed by direct interview with the patient or parent at the initial treatment visit and any other visit that occurs as part of routine care. At predetermined intervals between visits, the rater performs rating scales by telephone.
13. During the study, the treating physician does not see this research data. The patients and their parents are reminded that participation in the study should not interfere with routine clinical practice. Therefore, concerns about efficacy or side effects should be “called in to the office” per routine. To prevent introducing bias, results from the blinded rater will be sealed or available only to a data safety monitoring board until the trial is complete.
14. To reduce possible clinician bias in treatment decisions, guidelines for increasing, decreasing, and discontinuing medications consistent with routine clinical care are specified in advance.
15. Deviations from predetermined guidelines and reasons for doing so are recorded in the study record.
16. Patients and their families may decide to discontinue medication at their discretion or in consultation with the physician, as is done in routine clinical care, even if treatment guidelines do not suggest this. Patient decision or parent decision and reason(s) for discontinuing medication are recorded in the study record. In countries without universal health care, the reason might include cost or lack of health insurance.
17. Pill counts or other compliance measures are performed at each visit.
18. Outcomes of interest (e.g., tic symptom severity scores) are analyzed with mixed model, repeated measures regression. Use of pretreatment symptom severity data, including duration of collection prior to intervention, would likely be a covariate in the model, with specific details contingent upon the particular study hypotheses.
19. Duration of compliant treatment as a separate outcome is compared using survival analysis. This is based on the idea that patients who perceive a treatment to be effective are more likely to comply and continue treatment. Therefore, significantly longer adjusted duration of treatment for one arm would be a surrogate indicator of greater effectiveness. In some cases, patients may discontinue medications because they believe their symptoms no longer require treatment. When this occurs, longer duration of treatment no longer indicates treatment benefit. For subjects identifying this as a reason for discontinuation, the survival analysis will have to be modified. Frequency of these events could be analyzed separately as proportions, compared across treatment arms.
Independent, Blinded Rater and Performance of Clinical Rating Scales
In our experience with both clinical and epidemiological studies, careful training is essential to allow personnel to use these scales accurately. There is potential for diagnostic misclassification of potentially overlapping symptoms. For example, complex tics may resemble compulsions or nervous habits. In addition, families may refer to a wide variety of hyperkinetic behaviors as tics. Inaccurate and inconsistent classification of symptoms increases the likelihood of invalid results.
The blinded rater must be properly trained but does not need to be a physician. For studies involving ADHD treatment, rating scales from school teachers have been successfully employed,
16 and neurologists have trained raters for a school-based epidemiological study
33 and a clinical-neurophysiological study.
34 A consistent rater for each individual throughout a study is ideal, and the same rater should evaluate patients on two or more treatments. Ambiguous symptoms may need to be observed directly. During the prestudy training period, agreement between a physician and nonphysician rater should be determined using intraclass correlation coefficients for continuous measures (scales) and kappa statistic for categorical variables.
Potential Statistical, Ethical, Safety, and Economic Issues
Issues Related to Randomization.
In general, the benefits of randomization are preserved in this trial design. Randomization of subjects into treatment groups reduces the likelihood that a difference in treatment outcomes is due to preexisting characteristics of the patients receiving the treatments. In a study where a large number of variables are present, some may be unbalanced, even across randomized groups. Standard statistical theory would classify any recognized and measured unbalanced factors as covariates in the analysis.
Issues Related to Blindedness.
Some of the benefits of blindedness are preserved in this study design. The primary outcomes for statistical comparison are scored by a blinded rater. However, the patients are not blinded to their treatment status because they obtain their medication at the pharmacy.
For active comparator trials, the probability that preexisting beliefs would bias outcomes reporting in subjects who agree to participate is unknown and likely depends on the availability of information about the specific treatments. Because families often read about medications on the Internet or from direct marketing materials from pharmaceutical companies, some biased reporting might occur. However, it should be noted that this problem may also apply to those who participate in a standard, double-blind trial as well. If a patient’s family has acquired a prestudy belief that one medication is bad, they may refuse to participate in a study in which there is a chance they could be randomized to that treatment. One partial solution to this problem for all types of studies in which certain medications may be prejudged, involves the presence of a trusting doctor-patient relationship. When there is a good relationship with patients and there is genuine and perceived equipoise in an active comparator trial, careful explanation to the subjects may possibly reduce responder bias and decisions about nonparticipation, similar to the conversations that occur in clinics daily as part of routine care.
Individuals with a strong belief in one of the study medications may choose not to participate. In such cases, those who agree to participate would less likely be biased. In some cases, information could be included from those who do not want to be part of the trial but take one of the medications by choice. This group could function as another comparison group since presumably they would show the effect plus any bias. Additionally, patients could be questioned prior to treatment assignment as part of the informed consent procedure about any preexisting beliefs regarding treatment A versus treatment B. Decisions not to participate after finding out treatment assignment should be recorded as a dropout, along with the reason given and in accordance with the Consolidated Standards of Reporting Trials (CONSORT) guidelines.
35The effects of unblinding the treating physician may also threaten the validity of the trial. This may be obviated in part by 1) delineating specific reasons, in advance, for dose adjustments and discontinuation; 2) the existence of perceived equipoise; and 3) the use of an independent, blinded rater.
Statistical Issues Related to the Use of Active Comparators in Place of Placebo.
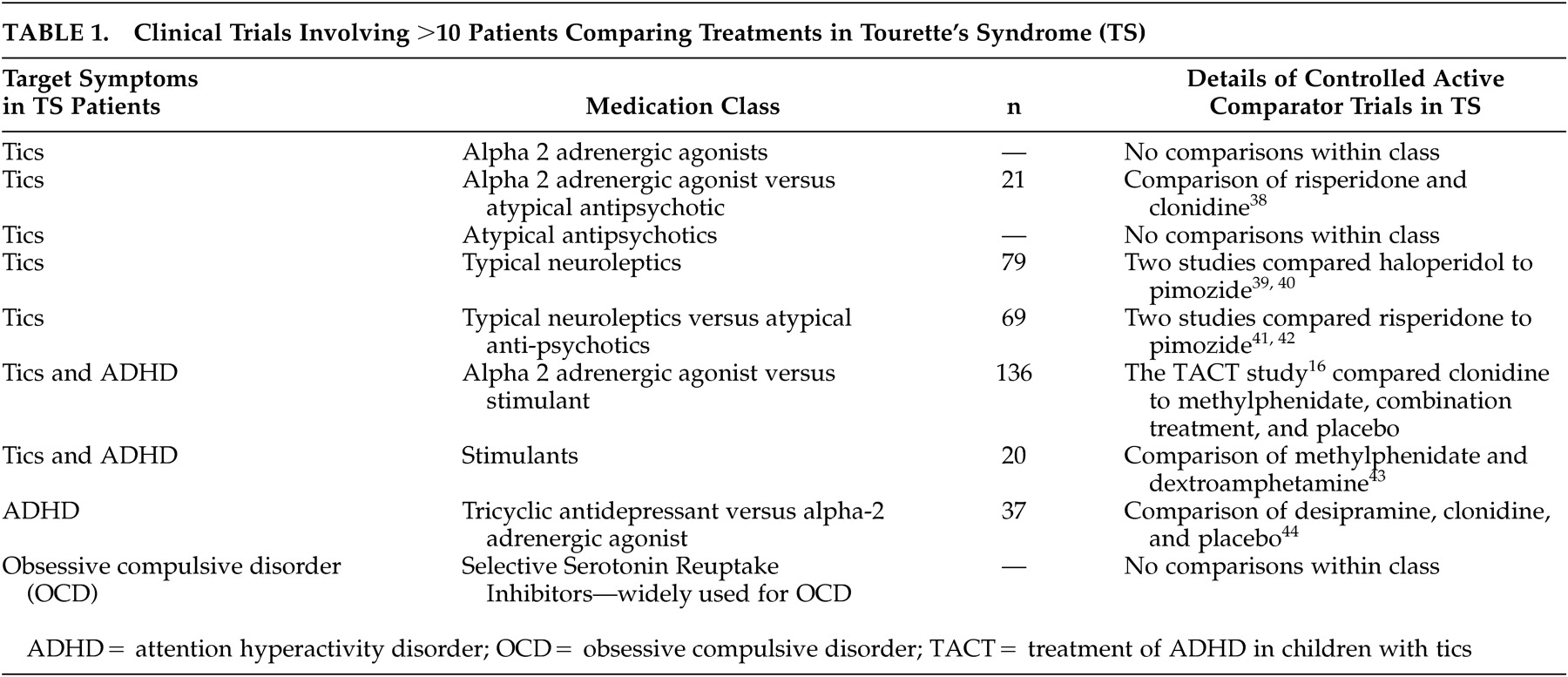
Active comparator trials have been performed successfully in TS (
Table 1). There are several issues related to the interpretation of outcomes. If an adequately powered, active comparator study shows no change in symptoms for treatment A or B at study endpoint, then one may reasonably conclude that there is no evidence of benefit from either treatment. However, if an adequately powered study shows that symptoms improve in both groups, but no difference is identified between treatments A and B, then it is difficult to disentangle the effects of time and natural waxing and waning of symptoms from the specific effects of medical treatment. In particular, it may not be clear whether A and B are equally effective or equally ineffective in the situation being studied. The apparently equal benefits at study endpoint may be due in both arms to nontreatment related factors (e.g., time).
However, this study design is proposed for drugs that are already marketed. Thus, in cases where the treatments have already been compared to placebo for this indication in this population and have shown statistically greater benefit, being equal does not pose new problems. Results in this study design for TS can and should be viewed in light of prior placebo-controlled clinical trials. In addition, we have proposed enrollment and collection of baseline data prior to any decision regarding treatment. Thus, a more prolonged assessment of prior symptom severity may provide a more accurate baseline for use in mixed models regression and reduce the likelihood that regression toward the mean will be misinterpreted as treatment-associated benefit.
Issues Related to Patient Safety.
Adverse events, particularly due to drug-drug interactions, may be more common in this type of study because patients are not as rigorously screened or intensively followed as they are in phase II and phase III single-issue studies and because many patients will be subject to polypharmacy. However, when clinical trials formally test what already occurs daily in clinical practice, the rate of adverse events should not exceed those in routine clinical practice, although the rates of detection of these adverse events may be higher. In addition, frequent rater-assessments by phone, which may occur in this type of study more often than clinic visits per routine medical care, may reduce health risks and costs.
36Issues Related to Conflicts of Interest or Coercion.
In current clinical research, investigators and sites are selected for multicenter NIH or industry sponsored trials based on a high volume of patients. Investigators often serve a dual role of investigator and treating physician, agreeing to participate in return for salary support and academic recognition. This may create a conflict of interest between the clinician-researcher’s best interest versus the patient’s best interest and open the door to coercive practices.
A randomized, open-label, active comparator study emulating standard practice may partially circumvent this conflict of interest. In this design, most investigator time will be reimbursed as per routine clinical care by third party payers. Unlike placebo-controlled trials, the care provided will not be substantially different than routine clinical practice, and therefore the decision to participate, made by the patient or the patient’s family, will not substantially change the care they receive. Less persuasion may be needed to convince subjects to enroll as well as to convince them of equipoise.
Issues Related to Costs of Research and Patient Costs.
Randomized controlled clinical trials cost thousands of dollars per patient. These costs are justified in phase II and phase III clinical trials, but this expensive paradigm is not required to advance evidence for medical decision making in all trials and works against the ability to generalize the results. The study design discussed here would essentially occur within routine clinical care, with extra funding needed only for administration, a study coordinator/rater, data entry, and statistical analysis.
Some patients without health insurance may choose to participate in research in order to have access to a medication they cannot otherwise afford. This poses a problem for the proposed design if medications are administered through routine care. The main difficulty may occur if active comparators include one drug that is substantially more expensive than the other, leading to higher dropouts on that arm. Reasons for dropouts, reported per the CONSORT guidelines, should make this problem transparent. However, it may be prudent to include this as a predictor variable in the study analysis if a high proportion of patients are uninsured. Alternatively, the study might be able to provide medication for those who could not afford it.
Summary
Once randomized controlled trials have supported safety and efficacy for a particular indication for a medical treatment, further questions often remain about long-term risks and benefits in the broad spectrum of patients who may have a disease but may not have been eligible for participation in phase III clinical trials. This issue is particularly important in neuropsychiatric conditions such as TS, in which polypharmacy is a common
37 yet largely untested practice. In addition, little or no rigorous evidence currently exists to guide treatment decisions between agents. We propose that randomized, rater (only)-blind, active comparator trials may yield valid, generalizable estimates of treatment effects in TS, complementing and enhancing the results of placebo-controlled trials. In the proper circumstances, these studies may be interleaved into routine clinical care with less expense than placebo-controlled clinical trials and with fewer conflicts of interest. They may also be ideal for obtaining long-term treatment data, especially in patients with comorbid conditions.
ACKNOWLEDGMENTS
This study was supported by NINDS/NIH grant K23 NS41920 (DG).