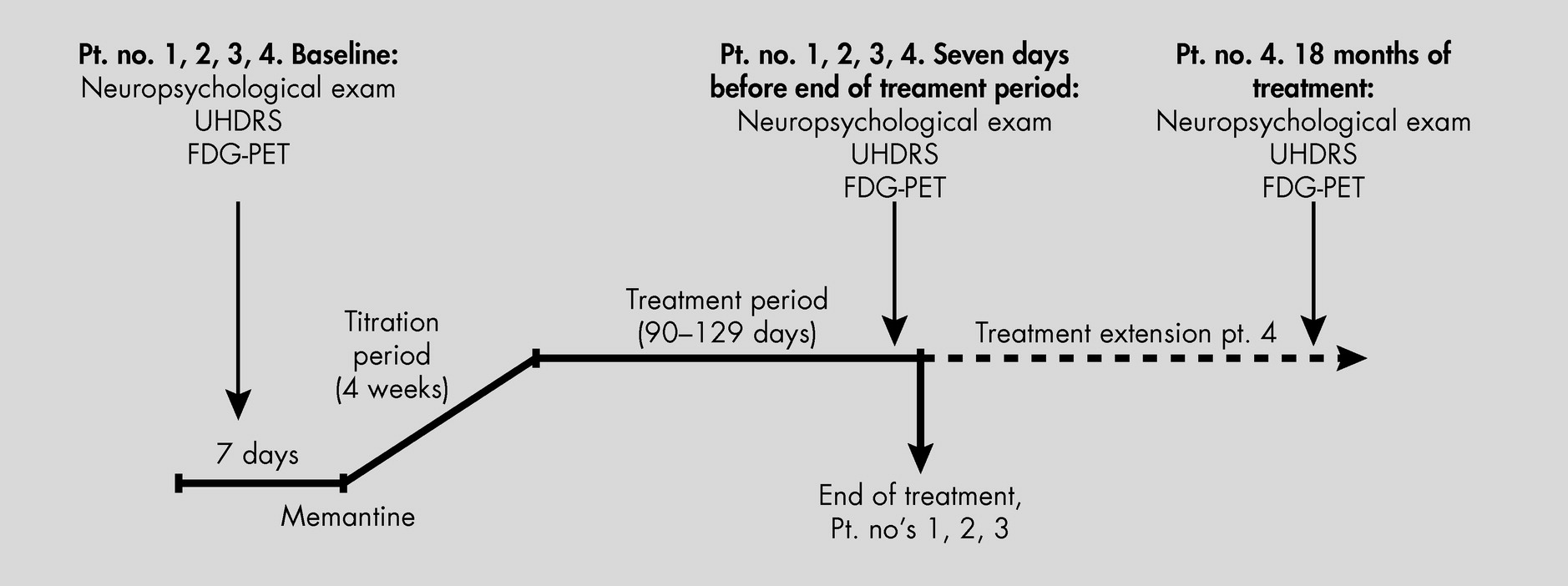
Treatment of HD, as well as reliable and reproduceable objective measurements to define progression of this devastating and heterogeneous disease, have been sought for years. In this trial, we united experience from earlier studies suggesting clinical effects of memantine on HD
4,5 and [
18F]FDG-PET-scans as a sensitive marker of disease-progression in other neurodegenerative disorders.
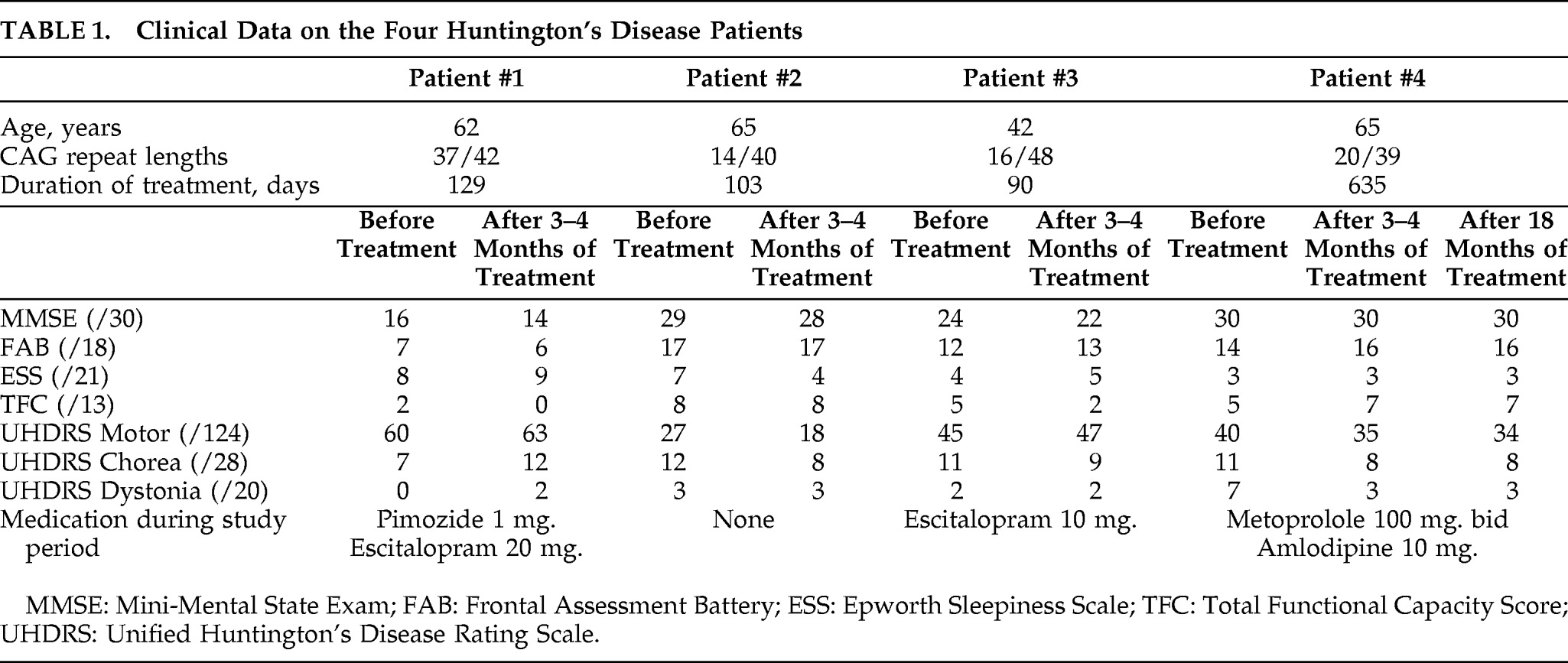
6,7 Treatment of four HD patients with memantine 20 mg/day for 3 months–4 months revealed no significant changes in the clinical and neuropsychological parameters. The patient with prolonged treatment did not deteriorate after 18 months, whereas the three patients treated for only 3–4 months all had progression at neuropsychological examination after 12 months. The relative FDG distribution, using PET, is an indirect marker of regional synaptic activity,
10 commonly used in the investigation of neurodegenerative diseases
11,12 and in earlier studies, a decreased metabolism is described before clinical changes, using [
18F]FDG-PET in pre-symptomatic mutation-positive persons.
6 We did not quantify the regional cerebral glucose metabolism in absolute physiological units; this would have involved repeated arterial cannulations, and it would have been an unacceptable trauma to most patients. Thus, in this investigation we can only observe changes in regional cerebral glucose metabolism relative to healthy-control subjects, and relative to other cerebral regions. Our data might represent a potential positive effect on the damaged synaptic transmission in the basal ganglia, as the cortical and striatal metabolism as assessed by [
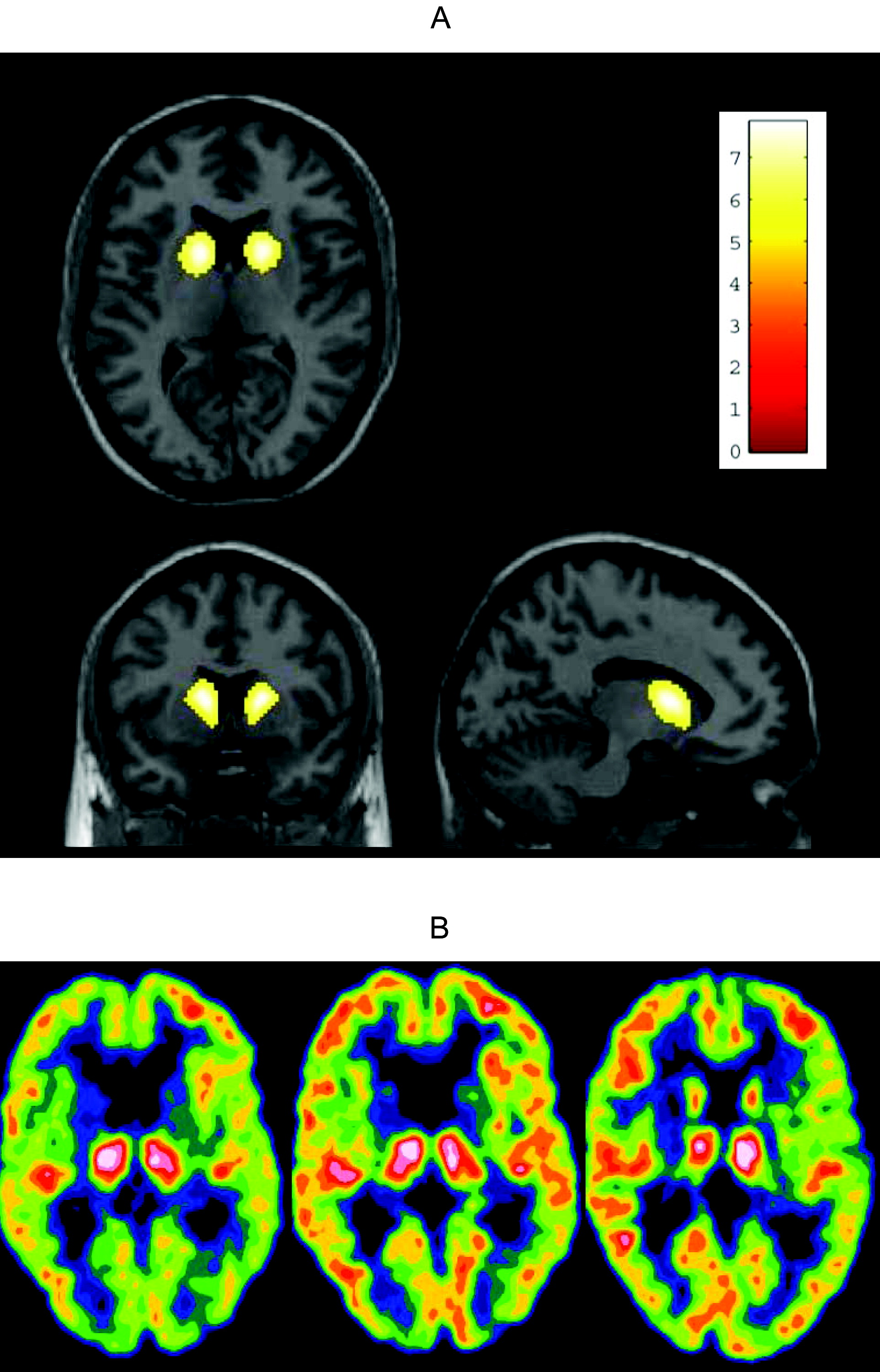
18F]FDG-PET was unchanged during the treatment period even in the patient who continued treatment and was scanned a third time after 18 months. Because this study did not include an untreated group of matched patients, we cannot estimate the natural cause of the relative regional cerebral glucose metabolism and can only hypothesize about the findings. From the literature, we would expect an average reduction per year of 2.3%–3.1% in caudate glucose metabolic rate.
13,14 Before treatment, there was relatively high thalamic activity relative to the cortex, which was reduced with treatment, to approach the value in the control group. This could be caused by either a reduction in thalamic metabolism or an increase in cortical metabolism. If the latter were the case, this would imply a relative increase in striatae; however, these statements are hypothetical interpretations of the trends in the data. This pilot study implies further limitations in the interpretation of the results: The hypothesized disease-modifying effect of memantine may be correct, but an acute pharmacological effect is another possibility. Furthermore, this study is too small to illustrate or exclude a tentative modifier effect of the patients' genotype besides the possible drug effect. Finally, a nontreated control group of matched HD patients would have been preferable. Treatment options are desperately needed, especially those with disease-modifying potential. This study can only provide preliminary data, and a long-term, randomized, double-blinded study design including [
18F]FDG-PET scans is highly desirable to further explore a potential positive effect of memantine.