Central pontine myelinolysis (CPM) is a clinically heterogeneous, demyelinating condition originally thought to occur only in the central pons.
7,8 When demyelination is found in areas outside of the pons, the disorder is referred to as either extrapontine myelinolysis (EPM) or central and extrapontine myelinolysis (CPEPM).
1–6 Identification of the importance of severe osmotic stress in the etiology of CPM led to the alternative name of osmotic demyelination syndrome (ODS). Disease severity varies from an incidental asymptomatic finding on imaging or autopsy to coma or death. Frequently, the symptoms of CPM are a combination of neuropsychiatric (e.g., emotional lability, disinhibition, and other bizarre behaviors) and neurologic (e.g., confusion, impaired cognition, dysarthria, dysphasia, gait instability, weakness or paralysis, generalized seizures).
9–11 The most common cause of CPM is an overly-rapid correction of hyponatremia in patients with conditions leading to nutritional or electrolyte stress, such as alcoholism, liver disease, immunosuppression after transplantation, malnourishment with underlying medical disease, gastrointestinal disease with acute electrolyte abnormalities, the syndrome of inappropriate secretion of ADH (SIADH), renal disease, cancer, pregnancy, and in high-endurance exercise (in triathletes, marathon runners, etc.).
1,4,12,13 Acute hyponatremia (≤48-hour development), as compared with a slowly progressive chronic hyponatremia (>48 hours in development), generally causes a more critical patient presentation, but more chronically developing hyponatremia can portend a higher risk for CPM.
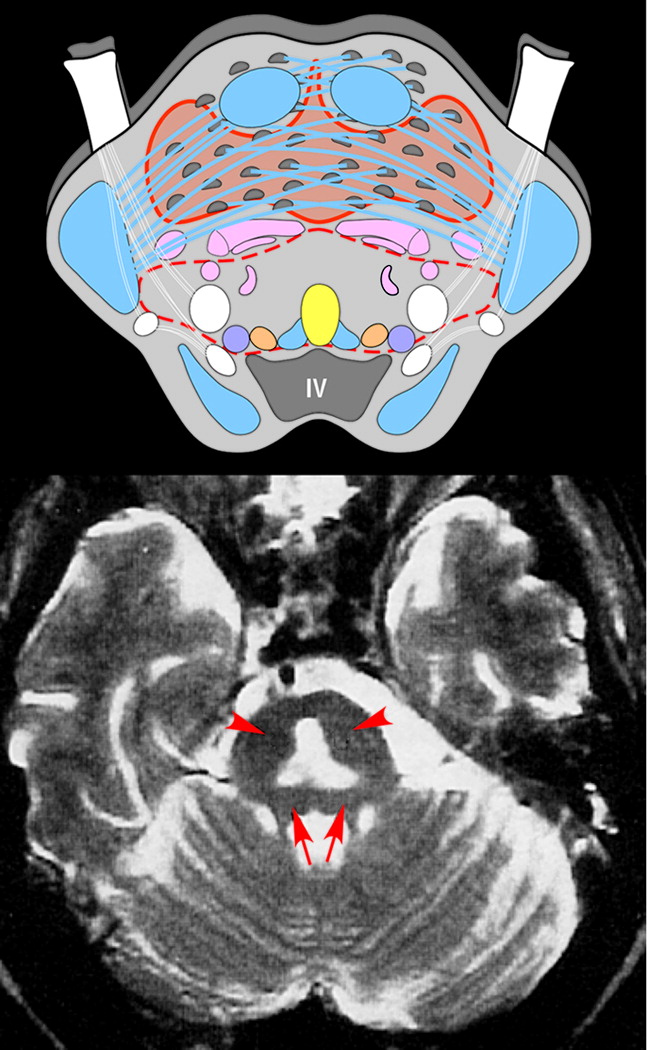
4,12,14The central pons is anatomically unusual in that gray matter and white matter are intermixed (
Figure 1), and this feature is presumed to account for the vulnerability of this area to osmotic injury. In most areas of the brain, oligodendrocytes are embedded within white matter and physically isolated from the capillary-rich gray matter.
15 It has been proposed that, in CPM, oligodendrocytes in close proximity to gray matter are exposed to a myelinotoxic substance as a result of osmotic stress. Although the mechanism of injury has not been definitely determined, both clinical and animal studies support the central role of osmotic stress, most commonly due to rapid correction of chronic hyponatremia.
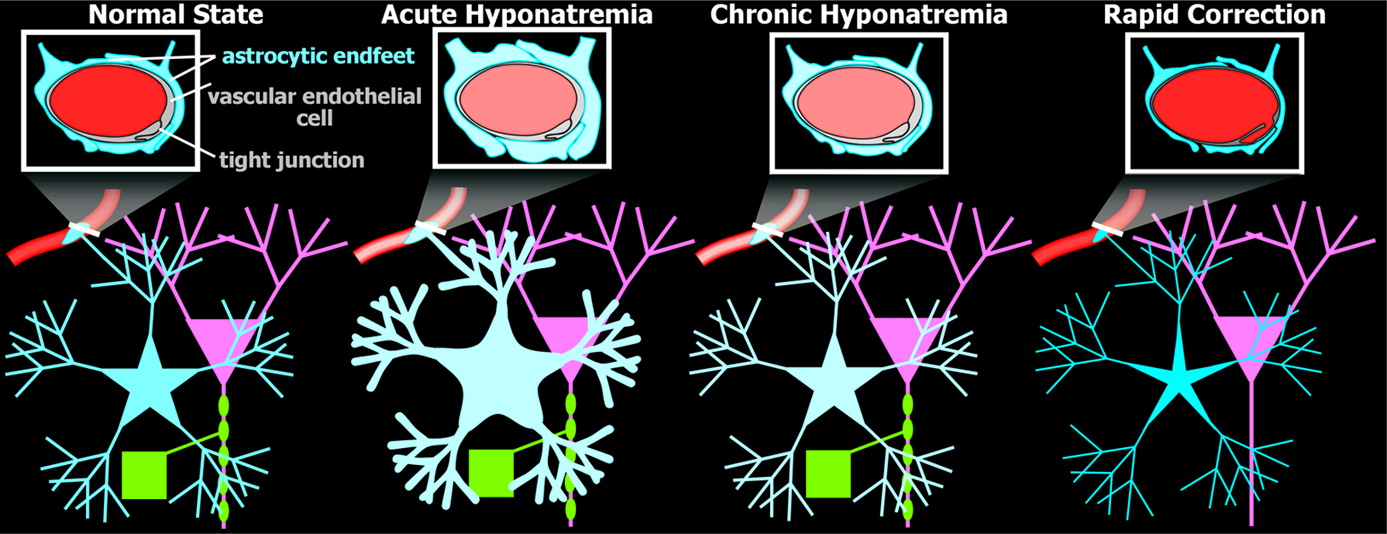
1,2,4 During acute onset of hyponatremia, water moves from the blood into the brain, causing brain cells (primarily astrocytes) to swell (
Figure 2).
2,5,6 Brain cells quickly adapt to hyponatremia by losing inorganic osmolytes (electrolytes) in order to reestablish normal cell volume. If the hyponatremic state is corrected during this acute phase, the brain can quickly accumulate electrolytes so as to restore normal osmotic equilibrium. Brain injury is likely to occur only in the most vulnerable patients (e.g., menstruating women, prepubescent children, patients with hypoxemia).
2 If the hyponatremic state continues, brain cells must also expel organic osmolytes (e.g., myo-inositol, taurine, glutamine) in addition to electrolytes in order to fully restore normal cell volume. Movement of organic osmolytes is much slower than movement of electrolytes. In the chronic hyponatremic state, brain-cell volume is normal, but the intracellular osmolality is low, in order for the brain to be in osmotic equilibrium with the blood (
Figure 2). At this point, correction of hyponatremia must be undertaken slowly in order to allow sufficient time for the brain-cells to reacquire both inorganic (electrolytes) and organic osmolytes. Osmotic stress due to rapid correction of chronic hyponatremia sometimes impairs blood–brain barrier (BBB) function, suggesting a loosening of the tight junctions between vascular endothelial cells (
Figure 2). This process may allow an injurious substance (e.g., complement) access to myelin. It has also been proposed that osmotically-stressed vascular endothelial cells release other substances (e.g., plasminogen activator, cytokines) that can injure myelin. An alternative possibility, suggested by the presence of structures similar to Rosenthal fibers within perivascular astrocytes (see below), is that myelin destruction is secondary to impairment in potassium siphoning, as has been recently proposed for several other demyelinating diseases.
2,16,17Several psychiatric illnesses (in addition to alcoholism) and many neuropsychiatric medications have hyponatremia as a potential complication. Over the past two decades, multiple case reports and case series have been published documenting hyponatremia and resulting CPM in patients with schizophrenia, anorexia, primary psychogenic polydipsia, and “ecstasy” abuse.
13,18–21 Among patients with chronic mental illness, 10%–25% may have primary polydipsia, increasing the risk of hyponatremia and premature death.
13,21 Primary medications or classes of medications used daily in neuropsychiatry in which hyponatremia is a side effect or reported to occur after use include carbamazepine, oxcarbazepine, the serotonin reuptake inhibitors (SSRIs), lithium, tricyclic antidepressants, opioids, and polypharmacy with multiple antipsychotic drugs.
20,22–24 Other classes of commonly used medications that increase risk of hyponatremia include salt-losing diuretics, nicotine, nonsteroidal anti-inflammatory drugs, and acetaminophen.
20CLINICAL COURSE
Historically, the prognosis in CPM or CPEPM has been considered very poor. More recent studies (primarily retrospective chart reviews), however, indicate a considerable range in both clinical course and prognosis in patients with clinically symptomatic CPM and/or CPEPM.
14,25–27 The first of these studies examined 44 cases, most (42/44) with chronic alcoholism.
25 Of the 32 patients with follow-up data who survived (2 died in the acute phase), 34% (11/32) had a “good outcome” (no significant functional deficits); 34% (11/32) had an “adequate outcome” (minor neurological deficits); and 31% (10/32) had a “poor outcome” (dependent). Another study in patients with alcoholism reported somewhat more favorable outcomes, with 44% (4/9) free of neurological deficits by 6 months after onset.
26 The authors noted that most of their cohort (8/9) did not have an episode of acute correction of hyponatremia. A more recent study examined 12 individuals with CPEPM related to a variety of medical causes.
14 In this more-diverse population, four patients died in the acute phase, and two were lost to follow-up. The remaining six were reported to have “good motor and cerebellar recovery.” However, all five of the patients who received neuropsychological testing had evidence of subcortical/frontal dysfunction, and most of these (4/5) were unable to return to work. The most recent study is also the only one to include prospective collection of objective follow-up measures of disability and functional status.
27 As in the previous study, this cohort was quite diverse in terms of causative conditions. Almost half (12/25) died either during the acute phase (2) or after hospital discharge (10). One was lost to follow-up. At final follow-up (mean 2.2 years; median: 1 year; range: 0–8 years), 29% (7/24) were normal; 17% (4/24) had mild cognitive or extrapyramidal deficits; and 54% (13/24) had a poor outcome (died or were dependent). Univariate analysis indicated that lower initial Glasgow Coma Scores, lower disability rating scales, lower sodium levels, and the presence of hypokalemia all predicted poorer outcomes. Although these studies differ in many respects, some consistent themes are emerging. A common pattern is an initial presentation with delirium, seizures, or encephalopathy, followed by a brief period (i.e., a few days) of lucidity or clarity before the onset of symptoms indicating CPEPM. These studies indicate that a “favorable” prognosis is possible, although few return to previous levels of functioning. The authors of one study made an interesting proposal, based on their results combined with data from previous publications, delineating three groups of patients with distinguishable prognoses.
14 Group One included patients who present with seizures and have a “good” prognosis. Group Two included those with cerebellar presentations (i.e., commonly, with severe alcoholism). These patients generally have good physical recovery, with residual cognitive impairment. Group Three comprised those with immunosuppression and liver transplantation. These patients generally present with more severe clinical features and do very poorly. This three-part grouping is in reasonable agreement with the prognostic factors noted above.
27The literature contains only a few reports of neuropsychological test results in cases of CPM and/or EPM. Two case reports included test results within 2 weeks of symptom onset.
28,29 A patient with only EPM (lesions in the basal ganglia) had severely impaired attention, verbal and visual memory, visuospatial function, frontal executive function, recognition memory, free recall memory, and naming, with preservation of other language-related functions.
29 All these deficits are consistent with previous reports in patients with basal ganglia lesions. In the other case, the patient had CPEPM (lesions in the pons, caudate, lentiform nucleus, thalamus, and internal capsules).
28 At 1 week, the patient had prominent deficits in attention and concentration (e.g., high distractibility, slow visual scanning), memory (immediate verbal recall and memory for daily events), visuomotor functioning, and fine motor speed. On reexamination at 4 months, the patient had improved somewhat in some domains, but deficits were still clearly present, and new problems included learning of new verbal information. Although acute neurological and behavioral symptoms had resolved, a new vocal tremor was present. Two other studies included neuropsychological test results obtained 1 month or more after symptom onset. A case report of CPM after liver transplantation noted lower-than-expected intellectual abilities, impairments of attention (auditory, divided, and selective), learning, problem-solving, and ability to use feedback to alter action, as well as pathological crying and laughter at 6 months after symptom onset, all consistent with a brainstem process.
30 Finally, in a case series described above, follow-up of five patients included neuropsychological testing (range: 1–19 months after onset of symptoms).
14 Although details of measures and scores are not provided, all five were noted to have subcortical/frontal dysfunction, and memory was impaired in two. Some of the patients in these studies had histories of alcoholism before CPM, possibly affecting the test results. Overall, although limited neuropsychological information is available in patients with CPM or CPEPM, many patients have lasting and significant cognitive impairments consistent with other pathological insults to the brainstem or subcortical structures.
PATHOLOGY
CPM was first documented in 1959, in patients with alcoholism.
7,8 The association with hyponatremia and rapid correction of the low sodium did not occur until many years later.
1–4 The defining neuropathological characteristics of CPM are symmetric, sharply demarcated lesions of the central pons, with selective destruction of myelin sheaths and loss of oligodendrocytes, presence of fat-laden macrophages, and absence of inflammatory infiltrate.
7,8 Blood vessels are patent, and both axons and neuronal cell bodies are generally spared. Swollen myelin sheaths and fragmented myelin are present in border regions. Centrally-located hypertrophic astrocytes are only lightly labeled for glial fibrillary acidic protein (GFAP), and GFAP
+ structures similar to Rosenthal fibers may be present within astrocytic processes around blood vessels.
16 In older lesions, there may be a central area of cavitation in which all elements have degenerated.
7,8,16 Evidence of BBB disruption (e.g., green discoloration in the pons in patient with jaundice) may also be present.
4 Pathological findings in EPM are similar to those of CPM.
1–4NEUROIMAGING
The appearance of CPM on computed tomography (CT) and MRI has been well characterized.
31–36 Standard clinical imaging can be normal for a considerable time (several days to 2 weeks) after onset of the clinical symptoms. After this period, the classic imaging findings of CPM generally emerge, and MRI is more likely to be positive than CT. Lesions are bright on T
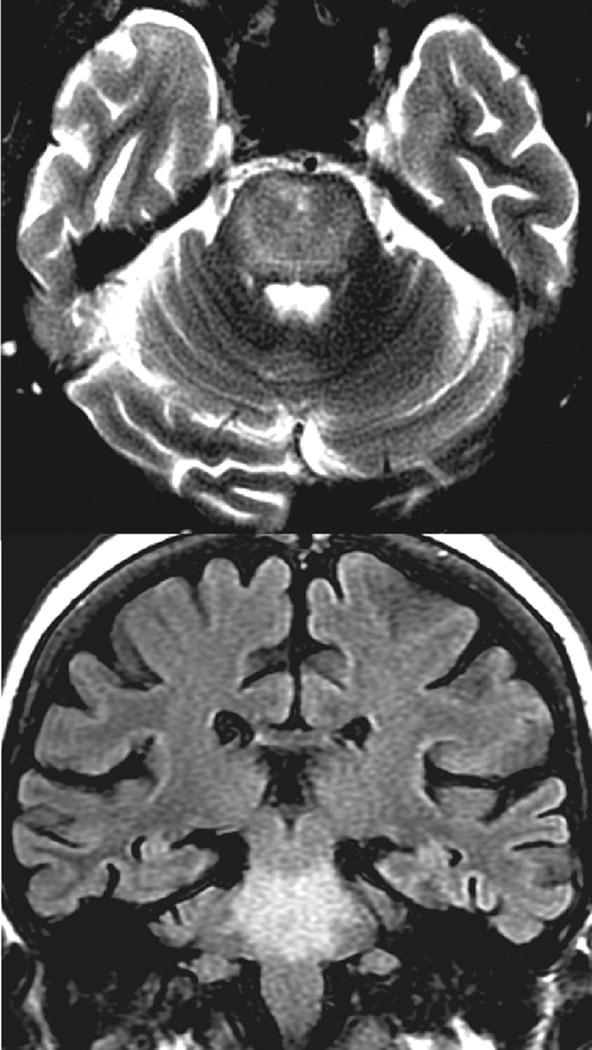
2-weighted (including fluid-attenuated inversion recovery, FLAIR) MRI sequences. Sparing of the ventrolateral pons, tegmentum, and corticospinal tracts results in the characteristic “trident-shaped” or “bat-winged” appearance (
Figure 1). The lesion is hypointense on T
1-weighted MRI, and there is no mass effect. Contrast enhancement is not usually seen. When present, enhancement will be noted in border regions.
37 If extrapontine lesions are present, they are often bilateral, and the most common locations are the cerebellar peduncles, globus pallidus, thalamus, lateral geniculate body, and putamen.
31,38 In many cases, the severity on imaging does not correlate with clinical manifestations.
37 However, as the clinical symptoms resolve, the T
2-weighted or FLAIR images may demonstrate a decrease in lesion size and intensity.
36,37,39 The differential diagnosis of pontine lesion on imaging includes pontine glioma, acute disseminated encephalomyelitis, vasculitis, hypertensive encephalopathy, multiple sclerosis, pontine infarct, and secondary radiation changes.
33,40,41Diffusion-weighted (DW) MRI is a newer pulse sequence that is very sensitive to changes in water motion associated with cellular dysfunction and so may hold promise for early diagnosis of CPM.
36 Multiple case reports and case series have recently described the presence of hyperintense pontine lesions with DW MRI in the first few days after symptom onset, when conventional MRI is usually negative.
31,37,41–44 Calculation of the apparent diffusion constant (ADC) at various time-points after symptom onset indicates that it is usually low early after symptom onset, then gradually rises to either a normal or somewhat elevated value. Lower-than-normal ADC values are associated with cytotoxic (intracellular) edema, and higher-than-normal ADC values are associated with vasogenic (extracellular) edema.
42 The recent demonstration that ADC values are modulated in-vivo by the level of aquaporin expression in astrocytes may also be relevant to CPM.
45 Some authors assert that hyperintense lesions on DW MRI associated with low ADC values help to differentiate CPM from the other pontine disorders described above (in the differential diagnosis).
37,41,42,44 It has also been proposed that if the ADC values are either normal in the presence of increased signal on T
2-weighted MRI, or clear to normal rapidly, the prognosis for recovery is better.
37,46 These preliminary results are quite promising, but larger studies are needed to fully understand the clinical benefits in using DW and ADC values in evaluation of the CPM patient.
Only a few studies have used advanced neuroimaging such as positron-emission tomography (PET), single photon-emission computed tomography (SPECT), and MR spectroscopy (MRS) in patients with CPM or CPEPM. One case report of PET examination in a comatose patient with CPEPM reported hypermetabolism in the acute phase (1 week after symptom onset) in the right temporal cortex, thalami, and pons.
38 By 1 month, the pontine and thalamic areas were hypometabolic, with the right temporal cortex remaining hypermetabolic. The authors speculated that hypermetabolism might be secondary to pathological microglial and/or astroglial activity. Two case reports of striatal EPM have reported decreased dopamine transporter binding (presynaptic) as measured by SPECT, suggesting osmotic injury to the nigrostriatal dopaminergic system.
47,48 In one of these cases, postsynaptic dopamine D
2 receptor binding was also assessed and found to be decreased.
48 Both cases responded well to carbidopa/
l-dopa treatment. Post-treatment resolution of both binding abnormalities was confirmed in one case.
48 The authors noted that the initial decreased ligand uptake and subsequent lesion resolution in the basal ganglia clarified the pathology of EPM and correlated with clinical recovery. Changes in proton MRS have been reported in both hyponatremia and CPM. An early study comparing patients with chronic (>1 week) hyponatremia with healthy individuals reported reductions in cortical levels of multiple metabolites, including choline (Cho), creatine/phosphocreatine (Cr), and
N-acetylaspartate (NAA), which returned to normal after clinical recovery.
49 The authors noted that their results are consistent with the proposal that many of these compounds are osmolytes. A recent study combined proton MRS and MR perfusion to examine the pons in a case of CPM after liver transplantation.
44 In the acute stage (8 days after onset), decreased NAA/Cr ratio, increased Cho/Cr ratio, and increased perfusion (as indicated by cerebral blood-volume mapping) were present. In the chronic stage (Day 48), there was further decrease in NAA/Cr and increase in Cho/Cr, and perfusion was decreased. The authors speculated that the acute spectroscopic findings were indicative of neuronal loss and gliosis, with the later findings related to further neuronal loss, but spared gliosis. More studies are required to better understand the functional significance of proton MRS changes, as multiple roles have been proposed for many of these substances.
50,51TREATMENT
Hyponatremia occurs when the serum sodium drops below 135 mmol/liter. Acute hyponatremia is not uncommon in several populations, including hospitalized patients and nursing home residents.
12,52 Chronic hyponatremia is more common in outpatient settings.
53 Treatment algorithms include considerations of current symptoms and risk for acute decline (to coma, respiratory arrest, or death), acuity of the hyponatremia, presence of chronic malnourishment, underlying medical illnesses or complications, volume status of the patient, and other accompanying electrolyte abnormalities. The choice between correcting hyponatremia with hypertonic 3% saline for brief periods versus much slower initial correction with normal saline requires a thorough clinical assessment and expertise in fluid and electrolyte balance. The overall goal is to achieve correction of serum sodium at a rate not exceeding 0.5 mmol/hour, or 0.5 meq/hour.
12,52,53 Frequent checks of serum sodium level are necessary during the stabilization phase. Conivaptan, a vasopressin V
1A/V
2 receptor antagonist, may be of assistance in treating acute euvolemic hyponatremia, as vasopressin is intimately involved in salt/water balance.
12 New interventions may be on the horizon for patients after CPM has occurred. Plasmapheresis and administration of intravenous immune globulins have been reported to improve outcomes.
54–56 Other potential treatments include steroids, symptomatic treatment for specific neurologic or neuropsychiatric symptoms (e.g., antiparkinsonian agents for movement disorders or psychiatric medications for agitation), and psychotherapies or behavioral interventions, if psychogenic polydipsia is present.
CONCLUSION
In summary, CPM or CPEPM are devastating and often preventable conditions, with considerable morbidity and/or mortality. Prevention is certainly the key, as current treatment only rarely leads to full recovery. Although imaging advances and a broader neuropathological understanding have improved diagnosis in the last few decades, CPM remains a condition for neuropsychiatrists to be aware of, particularly in view of the multiple neuropsychiatric medication classes in use and the psychiatric illnesses in which hyponatremia is a potential risk.