Almost all G protein-coupled receptors are tightly regulated by a common desensitizing mechanism. The process of agonist-specific, homologous desensitization of receptors is characterized by an increase in the refractoriness of a receptor to signal in response to repeated or sustained exposure to its agonist, limiting both the magnitude and the temporal extent of the receptor signal, thus protecting cells from overstimulation (for review, see reference
1).
Our knowledge concerning the basic mechanisms underlying the phenomenon of desensitization has been far advanced during the last decade. It involves the activities of two families of proteins: G protein-coupled receptor kinases and arrestins (for review, see reference
2). Following phosphorylation by G protein-coupled receptor kinases, G protein-coupled receptors bind to a family of soluble proteins named arrestins, which “arrest” G protein-coupled receptor signaling
(3–
5). Arrestins bind to regions of G protein-coupled receptors that are also primary determinants for G protein interaction, thus uncoupling G protein-coupled receptors from G proteins
(5). To date, four members of the arrestin gene family have been cloned
(6). Two arrestins, visual arrestin and cone arrestin, are expressed almost exclusively in the retina
(7), while β-arrestin-1 and β-arrestin-2 are ubiquitously expressed proteins. Although most of the research regarding the desensitization process has been carried out using the β2-adrenergic receptor as a model, it is now clear that this process regulates the function of many G protein-coupled receptors, including α- and β-adrenergic, muscarinic cholinergic, serotonergic, dopaminergic, angiotensin, and endothelin
(5,
8).
Antidepressants are well-known for their potential to modulate the density of functional neurotransmitter receptors such as β-adrenergic receptors in the brain
(9,
10) as well as in cultured cells
(11,
12). β-Adrenergic receptor down-regulation is accompanied by decreased receptor-stimulated cAMP formation
(13). The mechanisms for this reduction in receptor numbers are not completely understood. It is important to note that the onset of both down-regulation and clinical effectiveness require 10–20 days of antidepressant treatment
(14). It has been proposed that the reduction in the number of functional β-adrenergic and other receptors could be a regulatory response to the enhanced presence of the neurotransmitter in the synaptic cleft. The neurotransmitter’s elevated concentration is induced by acute inhibition of its reuptake or of monoamine oxidase activity by antidepressants
(9,
15). Some clinically effective antidepressants, however, neither influence norepinephrine or serotonin reuptake nor inhibit monoamine oxidase activity but still cause receptor down-regulation. Decreased monoamine receptor densities following antidepressant treatment can also be seen in cell culture systems lacking a presynaptic input
(12,
16). Thus, monoamine receptor down-regulation may directly result from postsynaptic actions of the antidepressants.
Biochemical research in mood disorders has focused, along the cascade of events involved in signal transduction, from studies at the level of the primary messenger (the monoamine neurotransmitter) to the level of the neurotransmitter receptors and lately to information transduction mechanisms beyond receptors involving the coupling of receptors with signal transducers (guanine nucleotide binding G proteins). Growing evidence suggests that G protein receptor coupling may be involved in both the pathogenesis and treatment of mood disorders (for review, see references
17,
18).
The existent knowledge concerning 1) antidepressant-induced receptor down-regulation; 2) delayed onset of both antidepressant-induced receptor down-regulation and clinical response to antidepressants, both requiring 10–20 days; and 3) involvement of signal transduction elements related to G protein receptor coupling in the pathophysiology of mood disorders, along with the emerging knowledge concerning the involvement of β-arrestins in the desensitization process and in uncoupling the G protein receptor interaction, led us to search for possible postsynaptic effects of antidepressant medications on β-arrestin levels and for possible alterations in β-arrestin levels in patients with a major depressive episode.
Method
Animal Study
Male rats (Sprague-Dawley, 250 g) were chronically treated intragastrically for 21 days with imipramine, desipramine, or fluvoxamine, 10 mg/kg, twice daily. Control rats were intragastrically treated with either distilled water or 10% ethanol. Rats given imipramine or desipramine (dissolved in distilled water) were compared with rats given distilled water. Rats given fluvoxamine (dissolved in 10% ethanol) were compared with rats given 10% ethanol. No significant differences were found between the two control groups of rats with regard to β-arrestin-1 levels. On the 22nd day, rats were decapitated, blood collected, and brain regions immediately dissected.
Rat brain tissue homogenization was carried out in a glass-Teflon homogenizer with ice-cold buffer containing 20 mM Tris-Cl pH 7.4, 2 mM EDTA, 1 mM 1 dithiothreitol, and antiprotease cocktail (Sigma). After initial centrifugation at 800 g for 5 minutes, the supernatant was collected and further centrifuged at 48,000 g for 30 minutes using a Beckman Ti80 rotor. The resulting supernatant and pellet were separated; the supernatant fraction was collected and centrifuged at 120,000 g for 45 minutes, and the supernatant obtained was used for all measurements. The pellet was resuspended in the homogenization buffer and the pellet fraction obtained was used for all measurements.
Serum was prepared from blood collected at the time of decapitation and kept frozen for measurement of antidepressant levels. Mean imipramine and desipramine levels measured by fluorescence polarization immunoassay in a TDxFLx system (Abbott) were 215 ng/ml (SD=40) and 308 ng/ml (SD=48), respectively.
Human Study
All subjects (patients with major depression and healthy subjects) were evaluated with the Structured Clinical Interview for Axis I DSM-IV Disorders by at least two senior psychiatrists. Interrater reliability for the diagnosis of depression was 0.91. Inclusion criteria for both patients and healthy subjects were 1) 18 years of age or older; 2) good general health with no clinically significant systemic abnormalities and no major findings from a physical examination; 3) normal laboratory test results for renal, hepatic, hematologic, and thyroid function and normal electrocardiogram results; 4) no treatment with antidepressants for at least 10 weeks; and 5) willing and able to give informed consent. Patients were required to have both a current diagnosis of major depressive episode (per DSM-IV criteria) that had not yet been treated with antidepressant medication and a score of at least 21 on the 17-item Hamilton Depression Rating Scale. Subjects with scores <21 were also examined but were included as a separate group. Exclusion criteria for all subjects were 1) history or evidence of clinically significant physical disorders; 2) current existence or history of a clinically significant diagnosable major psychiatric disorder (other than a major depressive episode for the patient group); 3) current or recent treatment (within the past 10 weeks) with psychopharmacological or other medications; and 4) alcohol or drug dependency or abuse within the previous 12 months. After complete description of the study to the subjects, written informed consent was obtained for a 20–60 ml blood donation. In all cases, blood was drawn between 8:00 and 10:00 a.m. The Hamilton depression scale was administered before blood donation. The study was approved by the Institutional Review Board.
The group of 36 untreated patients with major depression consisted of 27 patients (14 female and 13 male; average age=41.9 years [SD=12.3, range=19–71]) with Hamilton depression scale scores ≥21 and nine patients (four female and five male; average age=41.1 [SD=12.5, range=21–69]) with Hamilton depression scale scores of 10–20, whose β-arrestin measures were also included for the correlation between β-arrestin levels and Hamilton depression scale score. Patients were examined before the initiation of treatment. The healthy volunteer group consisted of 32 subjects (18 female and 14 male}; mean age=39.2 years [SD=12.6, range=20–75]) from the staff and staff’s families of Ben Gurion University.
Mononuclear leukocytes were isolated from 50 ml heparinized fresh blood of adult donors using Ficoll-Paque gradient according to Boyum
(19). Cells were homogenized in 25 mM Tris-HCl, pH 7.4, 1 mM Mg+2, 1 mM EGTA, 1 mM dithiothreitol, and antiprotease cocktail (1:100) (Sigma). The cytolosic fraction (supernatant) was separated from the membrane fraction by centrifugation at 18,000
g for 20 minutes. Membranes were suspended in homogenization buffer, and both fractions were frozen at –80°C until assayed. Aliquots were taken for protein concentration determination using the Lowry assay.
Immunoblot Analysis
For both the rat and human studies, cytosolic or membrane fraction was thawed on the day of assay. Protein aliquots (10 μg total) were taken for protein separation by SDS-(10%) polyacrylamide gel electrophoresis. The resulting proteins were transferred to nitrocellulose paper by use of electroblotting apparatus. Blots were incubated overnight with a monoclonal antibody to β-arrestin-1 (Transduction Labs, Lexington, Ky.). The immune bands were detected using the Enhanced Chemiluminescence Western Blot Detection System (Amersham, Piscataway, N.J.) followed by exposure to Kodak X-Omat film. The range of linearity of the assay as related to the protein concentration was found between 2.5–20 μg membrane-protein. Peak heights of immunoreactive bands presented as arbitrary absorbance units were determined with a computer-assisted imaging system for semiquantitative measurements. In each blot, 10-μg rat cortical membranes run as a standard reference.
Statistical Analyses
Bonferroni t tests were used for the statistical analyses of antidepressant drug effects on β-arrestin-1 levels in rat brain for various treatment groups in comparison with a single control group applying Bonferroni corrections. For the human study, Pearson’s correlation was applied for calculating correlations between β-arrestin-1 levels and clinical parameters (i.e., Hamilton depression scale ratings and age as well as biochemical parameters like G protein levels). Mann-Whitney rank-sum test and Dunn’s tests were applied for nonparametric comparison between patient and healthy volunteer groups.
Results
Animal Study
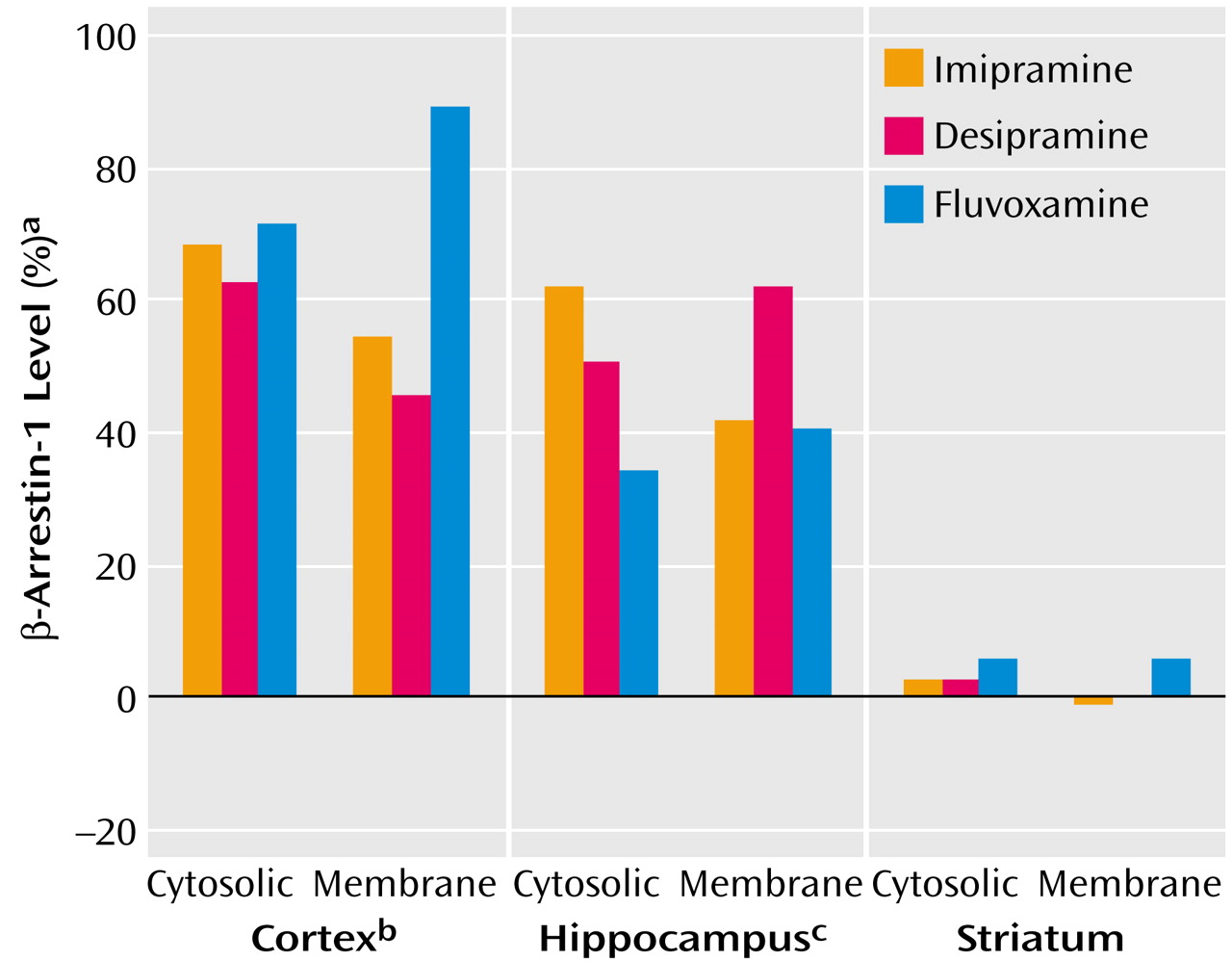
Rats were treated for 3 weeks with three types of antidepressants: the nonselective monoamine reuptake inhibitor imipramine, the norepinephrine-specific reuptake inhibitor desipramine, and the serotonin-specific reuptake inhibitor fluvoxamine. β-Arrestin-1 levels were measured in three rat brain areas: cortex, hippocampus, and striatum.
Figure 1 shows that both cytosolic and membrane β-arrestin-1 levels were significantly elevated by all three antidepressants in the cortex and in the hippocampus, while in the striatum no alterations in β-arrestin-1 levels could be detected.
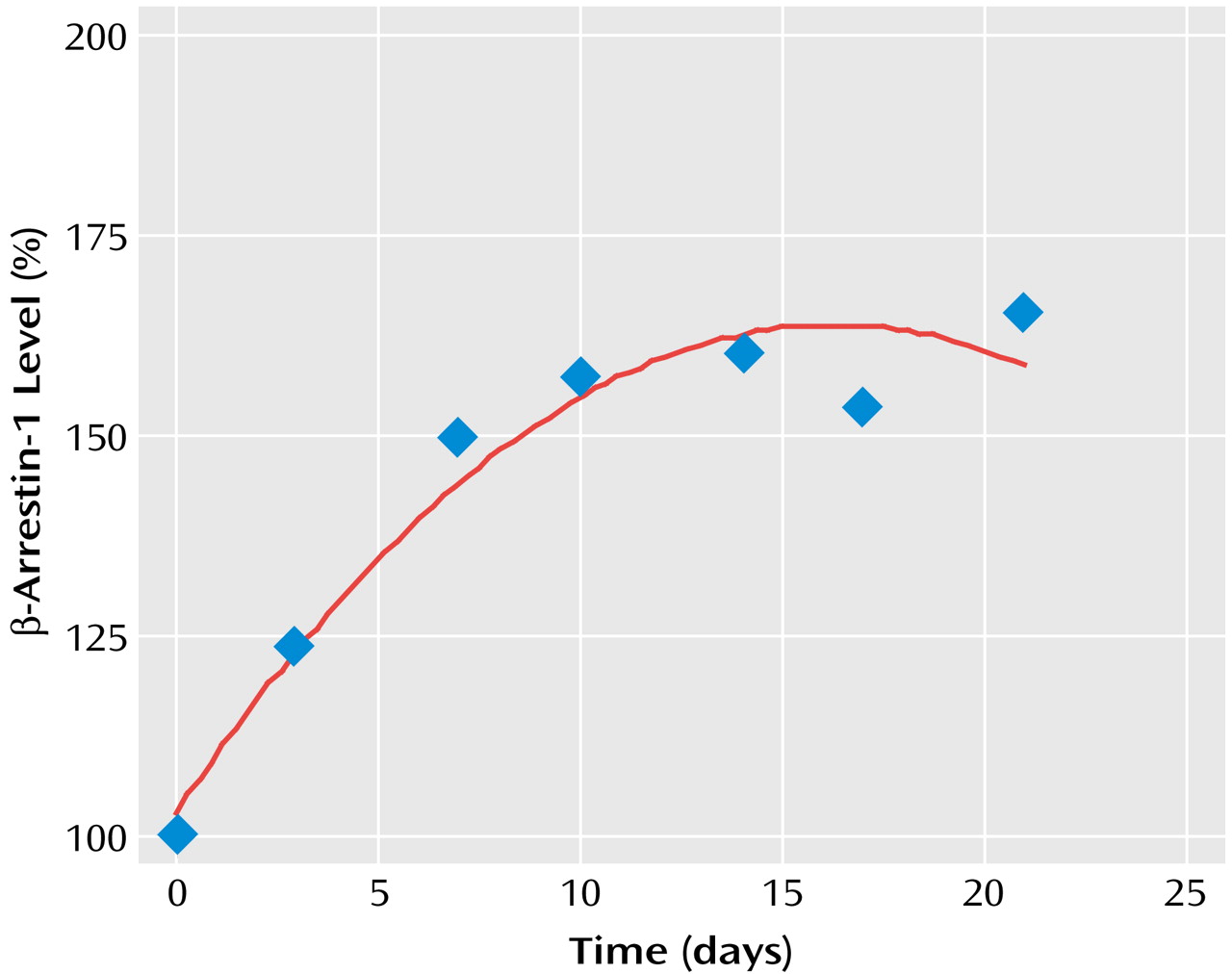
The dynamics of antidepressant-induced increases in the levels of both cytosolic and membrane β-arrestin-1 levels indicated that the difference became significant within 3 days and took 2–3 weeks to reach maximal increase (
Figure 2).
Human Study
The distinct elevations in the levels of β-arrestin-1 induced by antidepressant medications in rat brain areas suggested that alterations in β-arrestin-1 levels might exist in humans suffering from a depressive episode. Since it is known that β-arrestin-1 is expressed in mononuclear leukocytes, these cells were chosen for our human study. β-Arrestin-1 levels were evaluated in mononuclear leukocytes obtained from a group of patients diagnosed with major depressive episode (before the initiation of an antidepressant treatment) and a group of healthy volunteers. β-Arrestin-1 levels in the healthy volunteer group were independent of age (Pearson’s correlation coefficient=–0.06, p>0.50) or gender (average β-arrestin-1 levels for female and male subjects: 101.0% and 99.0%, respectively; Mann-Whitney rank-sum test: U=135, t=0.283, p>0.50).
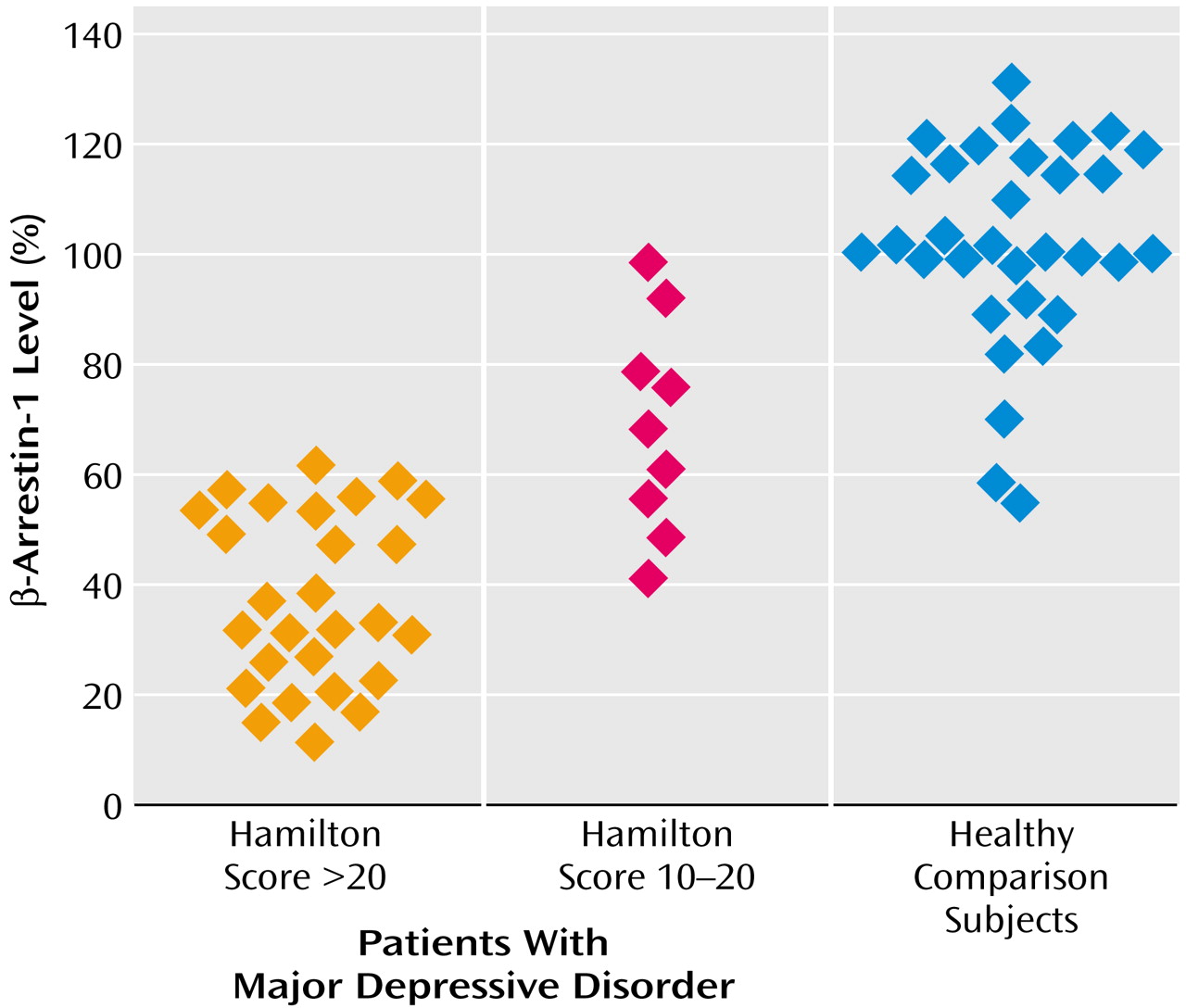
Figure 3 shows that β-arrestin-1 levels were significantly reduced in mononuclear leukocytes of patients with major depressive disorder (Hamilton depression scale score >20) in comparison with healthy volunteers. The sensitivity and specificity of the findings for diagnosing major depressive episodes were found to be 92.5% and 93.9%, respectively. Including patients with depression with ratings on Hamilton depression scale >16 still resulted in significant differences between the groups (Mann-Whitney rank-sum test: U=1134; t=6.86, df=67, p<0.0001) and high sensitivity and specificity values of 92.5% and 90.9%, respectively. The nine depressed patients with mild symptoms (Hamilton depression scale score=10–20) who were examined despite not satisfying the inclusion criteria (Hamilton depression scale score >20) also showed significantly reduced β-arrestin-1 levels in comparison with healthy volunteers. The extent of β-arrestin-1 reduction was significantly larger in depressed patients with scores >20 on the Hamilton depression scale than patients with mild symptoms (Hamilton depression scale score=10–20) (Q′=2.57, kappa=3, p<0.05, Dunn’s test) (
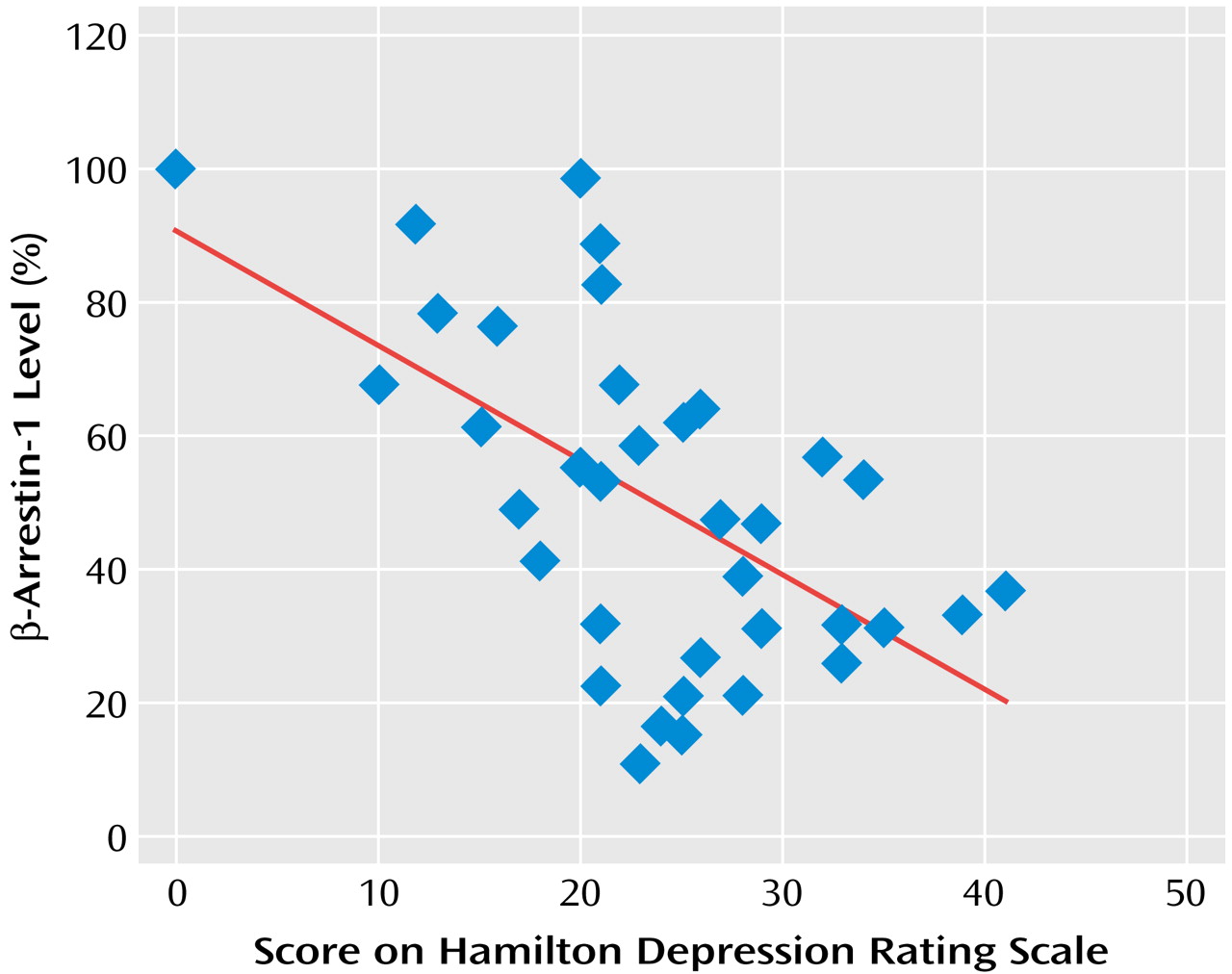
Figure 3). The degree of reduction in mononuclear leukocyte β-arrestin-1 levels was found to significantly correlate with the severity of the depressive episode as determined by the Hamilton depression scale score (
Figure 4).
Similar to our previous findings concerning reduced G protein levels in mononuclear leukocytes of patients with depression, the patients evaluated for G protein levels in the present study showed significant reductions in both Gα
s levels (mean=72.0%, SD=11.3%) and Gα
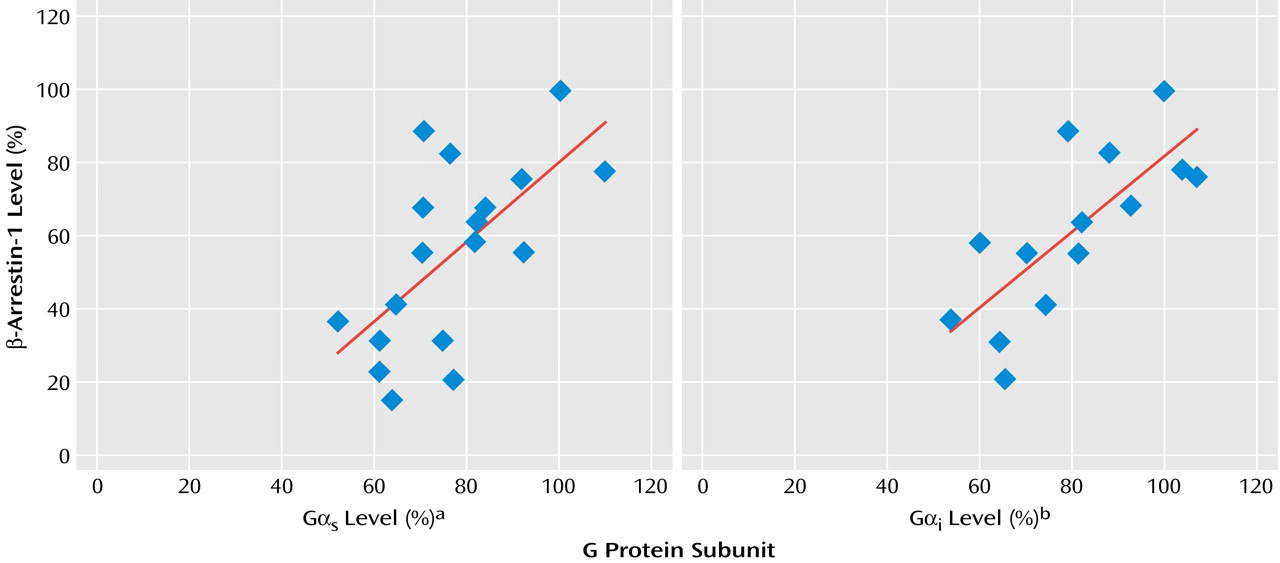
i levels (mean=73.2%, SD=12.0%). Cytosolic β-arrestin-1 levels correlated well with membrane G protein levels measured in the same mononuclear leukocyte preparation of the depressed patients (
Figure 5).
Discussion
Two types of studies are described in the present report: an animal study concerning the effects of antidepressant medications on β-arrestin-1 levels in rat brain and a human study concerning the levels of β-arrestin-1 in peripheral blood leukocytes of patients with depression. Preclinical and clinical findings were included in the same communication because they are both consistent with our major hypothesis concerning the importance of postreceptor desensitization regulators involving β-arrestin-1 to the pathogenesis, diagnosis, and treatment of mood disorders. One should not overlook the limitation that studies in naive animals may not reflect processes that happen in pathological tissue, and studies in peripheral cells are in fact distant markers of what occurs at relevant CNS synaptic sites.
A major finding of the present study was that antidepressant medications caused an elevation in β-arrestin-1 levels in rat brain. This process became significant within 3 days and took 2–3 weeks to reach maximal elevation. Another major finding was that β-arrestin-1 levels were significantly reduced in patients with depression. The extent of reduction of β-arrestin levels was correlated with the severity of the depressive episode.
Animal Study
The findings reported in the animal study suggest β-arrestin-1 as a possible biochemical underlying target site for the mechanism of action of antidepressants through receptor down-regulation and uncoupling or arresting G protein receptor signal transduction. The induction of β-arrestin-1 expression by antidepressants might be involved in the mechanism of action of various types of antidepressants: serotonin-specific, norepinephrine-specific, and nonselective reuptake inhibitors. The dynamics of antidepressant-induced increases in the levels of β-arrestin-1 in rat brain indicate that the process becomes significant within several days and takes 2–3 weeks to reach maximal increase. The time frame of antidepressant-induced increase in β-arrestin-1 levels correlates well with the time frame of antidepressant-induced β-adrenergic and other receptor down-regulation, as well as with the time frame of the clinical response. These findings lend further support to the clinical relevance of antidepressant effects on the expression of β-arrestin-1.
Increased levels of β-arrestin-1 may result in “arresting” intracellular signaling triggered by G protein-coupled receptors. Arrestin binding to receptors results in desensitization of G protein-mediated signaling by preventing interaction of receptors with G proteins. Antidepressant post receptor effects at the G protein receptor level are expected to show G protein receptor uncoupling because of the increased antidepressant-induced expression of β-arrestin-1 described in rat brain in the present study. Indeed, antidepressant-induced uncoupling of β-adrenergic G protein receptors
(20,
21) and 5-HT
1A G protein receptors
(22,
23) were previously described and are consistent with one of the classic biochemical hallmarks of chronic antidepressant treatment: down-regulation of several types of neurotransmitter receptors in the brain
(24). G protein receptor uncoupling induced by antidepressants through β-arrestin-1 elevation may also explain previous findings concerning antidepressant-induced switch of β-adrenergic receptor trafficking
(25), Gs redistribution
(26), and elevated Gs-adenylyl cyclase coupling
(27–
30).
Human Study
Functional and quantitative measures of G protein-coupled receptors detected in mononuclear leukocytes of patients were previously suggested as a possible biochemical diagnostic test for major depression
(31,
32). Comparing the reduced β-arrestin-1 levels with levels of Gα
s and Gα
i in mononuclear leukocytes of patients with depression, it is clear that the extent of reduction is more prominent by two- to threefold with respect to β-arrestin levels. While β-arrestin-1 levels were reduced by 62.9%, Gα
s and Gα
i protein levels were reduced by 28% and 27%, respectively, in unipolar outpatients with depression in the present study. Similar G protein level reductions have been observed by us in previous studies: Gα
s and Gα
i reductions of 21% and 23%, respectively, in unipolar depressed patients
(31); 29% and 39% in hospitalized depressed patients before the application of ECT
(33); 28% and 20% in seasonal affective disorder outpatients with winter depression
(34); and 21% and 17% for patients with bipolar depression
(35). Thus, it can be concluded that β-arrestin-1 measurements in mononuclear leukocytes of patients with depression might become a better diagnostic assay for detecting depression. Indeed, the sensitivity and specificity of the β-arrestin-1 test were found to be 92.5% and 93.9%, respectively. These values are far greater than the sensitivity and specificity values previously described for the immunoreactive G protein assay (73% and 81%). The fact that the extent of reductions in β-arrestin levels was found to correlate with the severity of depressive symptoms evaluated by the Hamilton depression scale score suggests that β-arrestin-1 reduction is probably a state-dependent marker. Thus, one would expect that patients recovering from depression under antidepressant treatment would show normalization in β-arrestin-1 levels in their leukocytes. Indeed, our preliminary (not shown) results suggest that β-arrestin-1 levels are normalized by antidepressant treatment in accord with the findings described in rat brain showing antidepressant-induced elevation in β-arrestin-1 levels.
An emerging view is that the binding of arrestins to heptahelical receptors also initiates a new set of signaling pathways in addition to blocking those mediated by G protein activation. The association of arrestins with heptahelical receptors does not simply uncouple receptors from G protein pathways but rather induces a switch in receptor signaling from classical second messenger-generating G protein-mediated pathways to other pathways such as those involving tyrosine kinases of the Src family, leading to activation of MAP kinase
(36). Moreover, arrestins have also been found to interact with a number of cellular proteins involved in endocytosis such as clathrin. Thus, arrestins may well represent multifunctional adaptor proteins that mediate a number of aspects of heptahelical receptor signaling. The findings described in the present study concerning the involvement of β-arrestin in the pathophysiology of major depression and in the mechanism of action of antidepressants may have extended implications concerning possible switch in postreceptor signaling related to tyrosine kinases, Src, and MAP kinase that may characterize major depressive disorder or may be induced by antidepressant treatment.