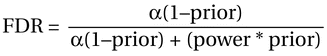Gene-by-environment interactions (G×Es) occur when the effect of the environment depends on a person's genotype or, equivalently, when the effect of a person's genotype depends on the environment. G×E research has been a hot topic in fields related to human genetics in recent years, perhaps particularly so in psychiatry. The first decade (2000–2009) of G×E research on candidate genes in psychiatry saw the publication of over 100 findings, many of them in top journals such as Science and the Journal of the American Medical Association. Such a large number of G×E studies in high-impact publications raised the prominence of G×E research in psychiatry and increased its appeal to scientists eager to build on past successes.
The excitement about G×E research also stems from its explanatory potential and the expectation that G×Es are common in nature. Genotypes do not exist in a vacuum; their expression must depend to some degree on environmental context. For example, genetic variants influencing tobacco dependence can have this effect only in environments where exposure to tobacco can occur. Similarly, G×Es could provide compelling explanations for why one person becomes depressed in response to severe life stressors while another does not (
1), or why cannabis use increases risk for psychosis in one person but not in another (
2). Indeed, it would be astonishing if G×Es did not exist, for this would mean that reactions to the environment are among the only nonheritable phenotypes (
3). Consistent with this expectation, twin analyses convincingly demonstrate that at least some responses to the environment are heritable (
4). Given these general reasons to expect that G×Es are common, most of the focus in psychiatric studies over the past decade has been on determining the specific genetic variants and environmental risk factors that underlie G×Es. In this article, we focus on such measured G×E studies as opposed to “latent variable” G×E studies, in which omnibus genetic risk is estimated using twins or other relatives.
The enthusiasm for G×E research has recently been tempered by increasing skepticism (
5–
7). Critics worry about the multiple testing problem combined with publication bias against null results (
6). The large number of potential G×E hypotheses—because of the many variables, operational definitions, and analyses that can be conducted—creates a large number of testable hypotheses, and there is a risk that only the “most interesting” (i.e., significant) findings will be published. To the degree that this occurs, the G×E literature contains an inflated number of false positives. Additionally, power to detect interactions is typically lower than power to detect main effects (
8), so the difficulties in detecting genetic main effects to date (
9,
10) may portend even more difficulties in detecting true interactions. Furthermore, interactions are sensitive to the scale on which the variables are measured (
11). Altering the scale (e.g., taking the logarithm of the dependent variable) can cause interactions to disappear, even so-called crossover interactions that are supposedly insensitive to scale (
7).
Perhaps most centrally, almost every G×E study conducted to date has used a candidate gene-by-environment interaction (cG×E) approach, whereby both genetic and environmental variables were hypothesized a priori. This is not an easy task given the inchoate understanding of the genotype-to-phenotype pathways in psychiatric disorders. Indeed, genome-wide association studies (GWAS) have largely failed to replicate reported associations from the candidate gene literature (
12–
14; however see Lasky-Su et al. [
15]). Thus, there is reason to question whether the candidate gene approach will be more successful in detecting replicable interactions than it has been in detecting replicable main effects.
Given such strongly polarized sentiments about cG×E research—excitement about the promise of cG×E research on the one hand and concern about the high rate of false positives on the other—we decided to survey the pattern of cG×E results in psychiatry in order to gauge whether there was evidence supporting the critics' concerns or whether the pattern of reported cG×E results was indicative of robust and promising findings. A formal meta-analysis across the entire cG×E field in psychiatry is not possible given the wide variety of interactions that have been examined. Nevertheless, by examining the patterns of cG×E findings, collapsed across the varied hypotheses investigated to date, we have attempted to gain some leverage on the state of cG×E findings overall.
Included Studies
We attempted to identify all cG×E studies published in the first decade (2000–2009) of cG×E research in psychiatry. We conducted searches using MEDLINE, PubMed, and Google Scholar, and we searched the reference sections of cG×E papers. Phenotypes in cG×E studies had to be DSM-IV diagnoses or closely related constructs (e.g., neuroticism). Only observational, as opposed to experimental, studies were included; pharmacogenetic studies were excluded. Studies were included only if there was variation across participants for phenotypic, genetic, and environmental variables (e.g., exposure-only designs were excluded).
In total, 98 articles encompassing 103 studies met inclusion criteria (five of the 98 articles reported results for two independent samples). A list of included and excluded studies and how they were coded is provided in the data supplement that accompanies the online edition of this article. Analyses were limited to interactions discussed in the abstracts of articles because results not mentioned in the abstract were often described in insufficient detail for accurate categorization. Each of the 103 studies was classified either as novel (containing no previously reported interactions) or as a replication attempt of a previously reported interaction. Replication attempts were defined as reports of an earlier cG×E finding in a separate article in which 1) the phenotypic variable was identified with the same name as the variable in the original report, even if specific scales differed (e.g., depression could be measured via self-report or clinician diagnosis); 2) the genetic polymorphism and genetic model (e.g., additive) were the same as in the original study; 3) the environmental moderator was substantively the same; and 4) replication results were reported for the same gender as the original report. Because of the inherent subjectivity involved in determining whether environmental moderators such as “stressful life events,” “maltreatment,” and “hurricane exposure” should be considered equivalent, we deferred to the primary authors regarding whether specific environmental variables measured the same construct. When possible, we report whether the original finding was actually replicated (p<0.05 in the same direction) for a given study. For example, Brummett et al. (
16) present significant results of a three-way interaction, but we used the clearly nonsignificant results of the two-way interaction that tested the original hypothesis (
17). When we could not clearly discern whether the original study was replicated, the replication attempt was excluded. Replication attempts were excluded for the following reasons: genetic model discrepancies (nine studies), gender discrepancies (eight studies), insufficient information (two studies), and replication attempt within the original report (one study).
Publication Bias Among Novel Reports of cG×E Studies
Publication bias, the tendency to publish significant results more readily than nonsignificant ones, is widespread in biomedical research (
18). While understandable given journal editors' motivation to publish findings with greater impact (typically novel, significant findings) and authors' decisions not to submit null findings (which require more work but have less payoff), publication bias is problematic because it produces a distorted representation of findings in an area of study (
19).
An indirect way to gauge the degree to which publication bias has occurred in novel studies (first reports of particular interactions) is to compare the rate of positive (significant) results among novel cG×E studies to the rate of positive results (that significantly replicated the original finding) among replication attempts. Replication attempts should more accurately reflect the true rate of positive cG×E findings because both positive and null replication results will be of interest to readers and be deemed publishable. Novel reports, on the other hand, may be deemed publishable only when positive. If so, publication bias will manifest as a higher rate of positive results among novel cG×E studies than among replication attempts. Consistent with this expectation, 96% (45/47) of novel cG×E findings were positive, but only 27% (10/37) of replication attempts were positive (Fisher's exact test, p=1.29×10–11). This p value should be interpreted with caution because many of the replication attempts were not independent of each other (e.g., the 5-HTTLPR-by-stressful life events interaction predicting depression was tested multiple times). Consequently we reran the analysis, excluding all but the first published replication attempt for each interaction. Despite the reduction in number of data points and the attendant loss of power, the results remained highly significant: 22% (2/9) of first replication attempts were positive, compared with 96% (45/47) of novel studies (Fisher's exact test, p=5.2×10–6).
These results are consistent with the hypothesis of widespread publication bias among novel cG×E reports, suggesting that many more tests of novel interactions have been conducted than reported in the literature. Given that increasing publication bias leads to an increasing field-wise type I error rate (because negative results go unpublished), these findings provide a clear warning against premature acceptance of novel cG×E findings.
Publication Bias Among Replication Attempts of cG×E Studies
The analysis above relies on the assumption that replication attempts provide a more accurate reflection of the true rate of positive cG×E findings than do novel studies. While probably true, publication bias may also exist among replication attempts themselves, meaning that less than 27% of replication attempts are actually positive. To test for evidence consistent with this possibility, we compared sample sizes of positive (significant and in consistent direction) replication attempts and negative (nonsignificant or opposite direction) replication attempts.
In the absence of publication bias, and when the hypotheses being tested are true, positive replication attempts should tend to have larger sample sizes than negative replication attempts because, holding effect size constant, larger samples provide greater statistical power (
20). This pattern of results—larger replication studies being more likely to be significant—occurs in fields where the relationships being tested have proven robust, such as the smoking-cancer link (
21). However, in the presence of publication bias, the opposite pattern of results could be observed—smaller replication studies may be more likely to be significant. This would occur if larger replication attempts were published irrespective of the direction of the results, whereas smaller studies were preferentially published when they yielded positive results. Consistent with the presence of publication bias among replication attempts (
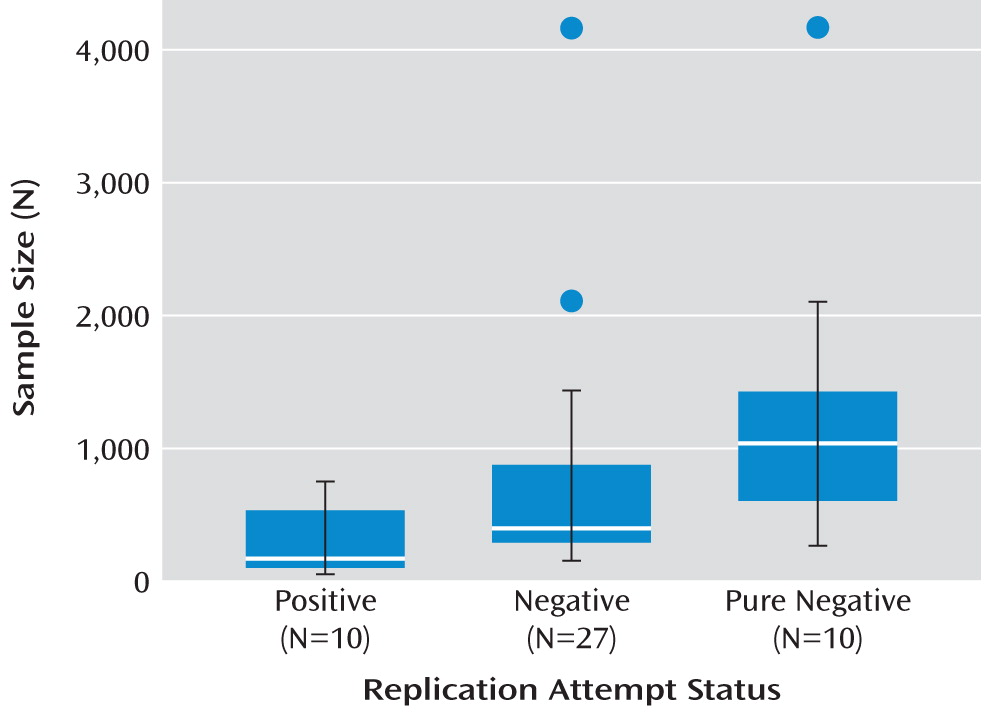
Figure 1), the median sample size of the 10 positive replication attempts was 154, whereas the median sample size of the 27 negative replication attempts was 377 (Wilcoxon rank-sum test, T=56, p=0.007). The nonparametric Wilcoxon rank-sum test was used because sample sizes were highly skewed, but results here and below were also significant using parametric tests.
We used one additional, independent approach to test for evidence consistent with publication bias among replication attempts, hypothesizing that negative replication attempts may be published more readily when reported with some other novel, positive cG×E finding. Consistent with this, 63% (17/27) of negative replication attempts were reported with novel, positive cG×E findings whereas only 20% (2/10) of positive replication attempts were published with novel, positive cG×E findings (Fisher's exact test, p=0.03). Moreover, it appears that much larger sample sizes are needed in order for negative replication attempts to be published: the median sample size of the 10 “pure negative” replication attempts (not published alongside another novel, positive cG×E finding) was 1,019, which is more than six times larger than the median sample size (N=154) of the 10 positive replication attempts (T=9, p=0.001; see
Figure 1).
Although publication bias is the obvious explanation for these otherwise counterintuitive findings, systematic differences between smaller and larger studies may also play a role. For example, Caspi et al. (
1) and Lotrich and Lenze (
22) argued that smaller cG×E studies tend to use higher-precision prospective measures, whereas larger studies tend to use lower-precision retrospective reports. If so, smaller replication studies may be more likely to be positive because they tend to analyze variables with less measurement error than larger replication studies and not because of publication bias. However, this argument does not explain why negative replications are published alongside novel cG×E findings more often than positive replications or why negative replications published alone have the largest sample sizes. Taken together, we believe that publication bias among replication attempts is the most parsimonious explanation for our results.
Power to Detect cG×Es
Statistical power is the probability of detecting a significant result given that the alternative (here, cG×E) hypothesis is true. Statistical power has been a central issue in modern psychiatric genetics, and it is likely that most candidate gene studies have been underpowered (
23). Several studies have likewise investigated the statistical power of cG×E studies (
24–
27) and have concluded that power to detect cG×E interactions is even lower, sometimes much lower, than power to detect genetic or environmental main effects. Low statistical power in a field is problematic, not only because it implies that true findings are likely to be missed, but also because low power increases the proportion of significant “discoveries” in a field that are actually false.
Interactions are tested by multiplying two first-order (here, gene and environment) predictors together, creating a product term. All three variables (the two first-order variables and the product term) are entered into the model, and a significant product term is evidence for interaction effect. It is often argued (e.g., in Caspi et al. [
1]) that the reduction in power to detect interaction effects is due to the correlation between the product term and the first-order predictors, but this is incorrect; the correlation between the product and the first-order terms plays no role in the power to detect interactions (
8). This can be seen by centering (subtracting the mean from) symmetrically distributed first-order predictors, which reduces the correlation between product and first-order terms to ∼0 but does not change the significance level of the product term. (The same effect occurs for nonsymmetrically distributed predictors, although the constant subtracted will not be the mean; see Smith and Sasaki [
28].)
The primary reason that power to detect interactions tends to be low is that the variance of the product term tends to be low in nonexperimental studies (
8). Power to detect the effect of any predictor, including a product term, increases as a function of the variance of that predictor. The variance of product (here, cG×E) terms is maximized when subjects are selected from the joint extremes (high G–high E, low G–high E, high G–low E, and low G–low E) of the two first-order predictors, but such jointly extreme observations tend to be rare in nonexperimental studies (
8). This issue is particularly relevant to cG×E studies, as it is generally not possible to sample from the genotypic extremes (e.g., equal numbers of the two homozygotes). Thus, power in cG×E studies will be maximized whenever variance in the two first-order predictors is maximized, that is, when the minor allele frequencies are high (e.g., 0.50 for biallelic loci) and when equal numbers of subjects are exposed to the extremes of the environmental moderator (
25). Additional factors such as ascertainment strategy (
29), study design (
30), correlation between the genetic and environmental variables (
8), and measurement error in the variables (
23) also affect statistical power to detect cG×E effects and should be considered in interpreting results from cG×E studies.
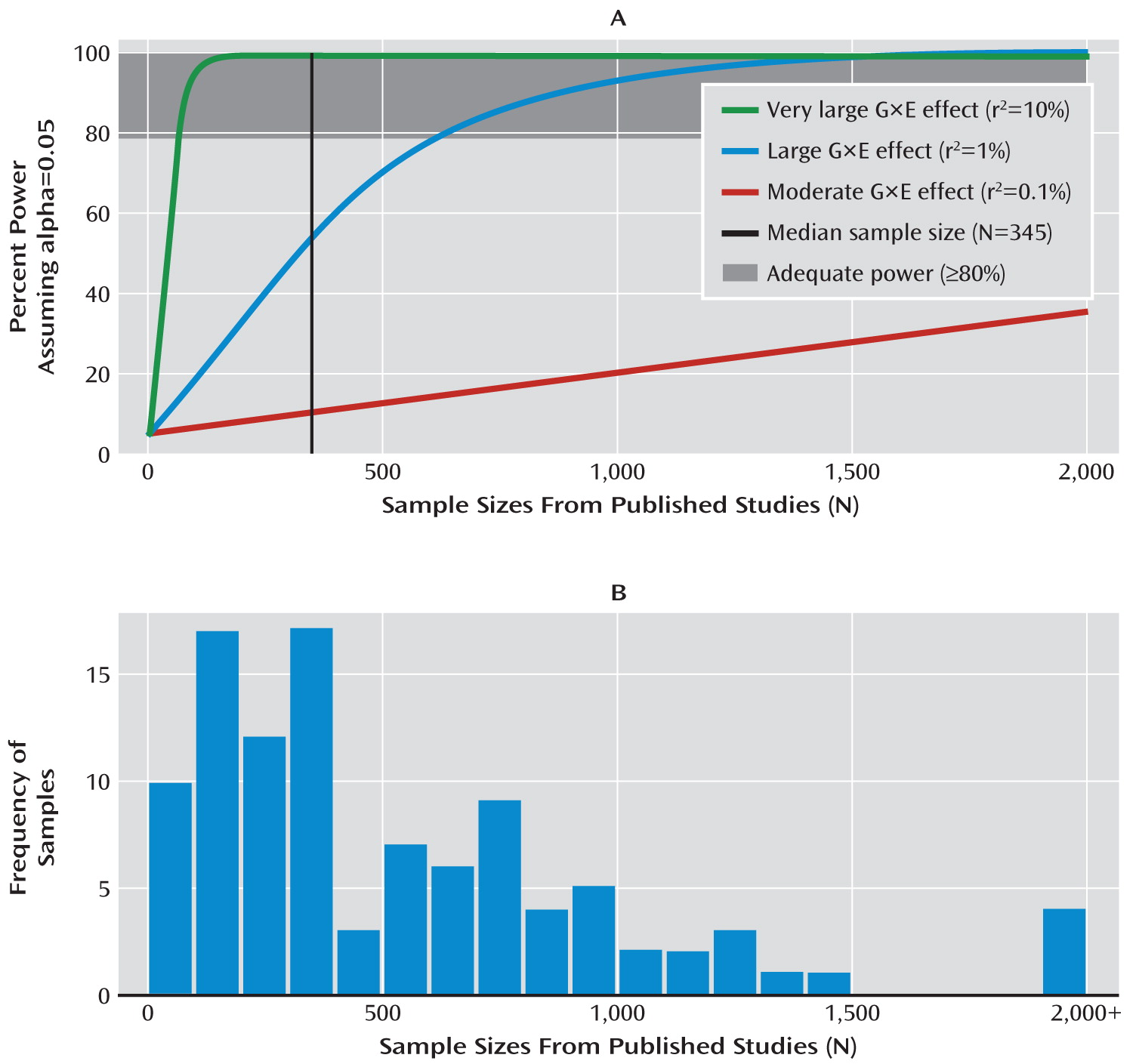
In
Figure 2A, we provide power estimates for cG×Es given three different effect sizes and plot them above a histogram of actual sample sizes from the first decade of cG×E studies (
Figure 2B). Power estimates were derived from 10,000 Monte Carlo simulations with alpha set to 0.05. We assumed that no error occurred in any of the measures and that the environmental and genetic variables accounted for 20% and 0.5% of the variance in the outcome variable, respectively. These are favorable values for the detection of G×E effects because increasing variance accounted for by the first-order terms increases power to detect an interaction term in linear regression.
In
Figure 2A, the three lines depict statistical power for three different possible cG×E effect sizes. As a point of reference, the effect size designations in
Figure 2A reflect what would be considered very large (r
2=0.10), large (r
2=0.01), and moderate (r
2=0.001) for genetic main effects in large GWAS, which provide the most reliable information about the true effect sizes of genetic main effects (
31). We used these effect sizes to provide points of reference, although it is possible that G×E effects tend to be larger or smaller than genetic main effects.
Sample sizes from the 103 cG×E studies are depicted in
Figure 2B. The median sample size, shown as a vertical line in
Figure 2A, was 345. Assuming a moderate effect size of r
2=0.001, statistical power was less than 10% for the median sample size. Given large and very large effect sizes, cG×E studies required sample sizes of ∼600 and ∼50 to reach sufficient statistical power (80%) to reject the null. In sum, unless cG×E effect sizes are over an order of magnitude larger than the typical genetic main effect sizes detected in GWAS, then cG×E studies have generally been underpowered, perhaps severely so, a conclusion also reached by others (
23,
32).
The False Discovery Rate in cG×E Research in Psychiatry
A necessary, albeit underappreciated, consequence of low power is that it increases the false discovery rate—the proportion of “discoveries” (significant results) in a field that actually represent type I errors (
33,
34). Other factors influencing the false discovery rate are the chosen type I error rate (typically α=0.05) and the proportion of tested hypotheses that are correct (the prior). Given these parameters, calculation of the false discovery rate is straightforward:
In addition to suggesting low power, candidate gene main effect research in psychiatry suggests that the priors in cG×E research may also be low. For one thing, candidate gene main effect studies in psychiatry have yielded no unequivocally accepted associations after more than a decade of intense efforts (
10), despite the fact that candidate gene main effect hypotheses were predicated on robust neurobiological findings. In contrast, GWAS have identified numerous replicable associations that have not usually been in candidate genes: out of 531 of the most robustly associated single-nucleotide polymorphisms (SNPs) to various medical and psychiatric phenotypes in GWAS studies, 45% were in introns, 43% were in intergenic regions, and only 11% were in exons (
35), the typical hunting ground for candidate polymorphisms. Furthermore, when candidate polymorphisms have been examined among GWAS results, they have usually not demonstrated better than chance performance (
12–
15).
Thus, accumulating evidence suggests that our understanding of the neurobiological underpinnings of psychiatric disorders has, to date, typically been insufficient to lead to correct hypotheses regarding candidate polymorphisms. Colhoun et al. (
9) estimated that 95% of candidate gene main effect findings were actually false positives, which translates to a prior of between 0.3% and 3% (assuming statistical power is between 10% and 90%). Because of the need also to specify the correct moderating environmental variable, generating cG×E hypotheses that prove correct may be even more difficult than generating (simpler) genetic main effect hypotheses. Thus, the prior for cG×E studies may be lower than the 0.3% to 3% it appears to be for candidate gene main effect hypotheses.
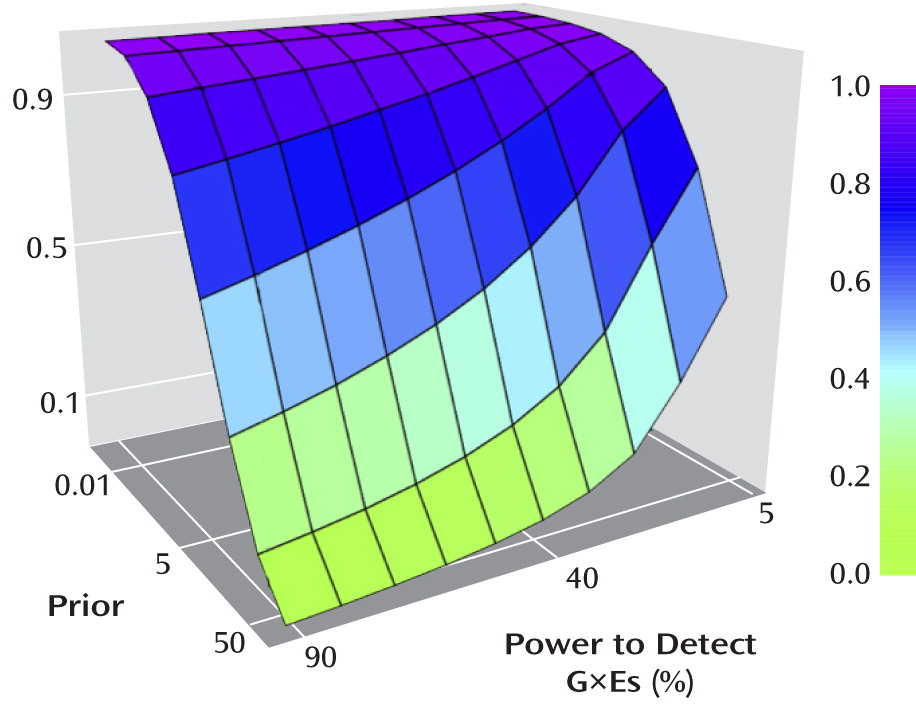
Figure 3 shows the false discovery rate as a function of varying assumptions about power and the prior. If cG×E hypotheses prove to be like candidate gene hypotheses, with (optimistic) values of the prior and power of 5% and 55%, respectively, then approximately two-thirds (63%) of positive findings would represent type I errors. Using values of the prior (1%) and statistical power (10%) that may be more realistic, the false discovery rate is 98%. Obviously, the true false discovery rate in the cG×E field may be higher, lower, or in between these values.
The 5-HTTLPR-by-Stressful Life Events Interaction Example
In 2003, Caspi and colleagues (
17) reported an increasingly positive relationship between number of self-reported stressful life events and depression risk among individuals having more short alleles at the serotonin transporter (5-HTTLPR) polymorphism. Their study has been extremely influential, having tallied over 3,000 citations and a large number of replication attempts. We reviewed this specific cG×E hypothesis and the attempts to replicate it because it highlights the important issue of direct compared with indirect replications and because it potentially illustrates the issues surrounding publication bias and false discovery rates discussed above.
Both direct cG×E replications, which use the same statistical model on the same outcome variable, genetic polymorphism, and environmental moderator tested in the original report, and indirect cG×E replications, which replicate some but not all aspects of an original report, exist in the cG×E literature. Indirect replications might sometimes be conducted to help understand the generalizability of an original report (
1) and might in other cases be conducted out of necessity because available variables do not match those in the original report. However, it is also possible that in an unknown number of cases, a positive indirect replication was discovered by testing additional hypotheses after a direct replication test was negative. Sullivan (
36) showed that when replications in candidate gene association (main effect) studies are defined loosely, the type I error rate can be very high (up to 96% in his simulations). The possibilities for loosely defined, indirect replications are even more extensive in cG×E research than in candidate gene main effect research because of the additional (environmental) variable. Thus, we believe it is important that only direct replications are considered when gauging the validity of the original cG×E finding (see also Chanock et al. [
37]). Once an interaction is supported by direct replications, indirect replications can gauge the generalizibilty of the original finding, but until then they should be considered novel reports, not replications.
The decision of how indirect a replication attempt can be in order to be included in a review or meta-analysis is critical for gauging whether a finding has been supported in the literature. With respect to the interaction of 5-HTTLPR and stressful life events on depression, a meta-analysis by Munafo et al. (
38) and subsequent meta- and mega-analysis by Risch et al. (
5) examined results and/or data from 14 overlapping but not identical replication attempts and failed to find evidence supporting the original interaction reported by Caspi et al. (
17). However, a much more inclusive meta-analysis by Karg et al. (
39) looking at 56 replication attempts found evidence that strongly supports the general hypothesis that 5-HTTLPR moderates the relationship between stress and depression. Karg et al. argue that these contradictory conclusions were mainly caused by the different sets of studies included in the three analyses. Karg et al. included studies that Munafo et al. (
38), Risch et al. (
5), and we, in this report, consider to be indirect replications. For example, Karg et al. included studies investigating a wide range of alternative environmental stressors (e.g., hip fractures), alternative outcome measures (e.g., physical and mental distress), and alternative statistical models (e.g., dominant genetic models). Furthermore, 11 studies included in the Karg et al. analysis used “exposure only” designs that investigate only those individuals who have been exposed to the stressor. We excluded such designs in this review because they do not actually test interactions; rather, interactions must be inferred by assuming an opposite or no relationship between the risk allele and the outcome in nonexposed individuals. Additionally, the result from at least one of the studies deemed supportive of the interaction in Karg and colleagues' meta-analysis (
40) is actually in the opposite direction of the original finding when the same statistical model employed in the original report is used (
5). Taken together, the pattern of results emerging from these three meta- and mega-analyses is surprisingly consistent: direct replication attempts of the original finding have generally not been supportive, whereas indirect replication attempts generally have.
There also appears to be evidence of publication bias among the studies included in the Karg et al. (
39) article. As we have shown to be the case in the broader cG×E literature, larger studies included in the Karg et al. meta-analysis were less likely to yield significant results. A logistic model regressing replication status (significant replication compared with not) on sample size among studies included in their meta-analysis found that the odds of a significant replication of Caspi's original finding decreased by 10% for every additional 100 participants (β=–0.001, p=0.02).
Karg et al. (
39) touch on the possibility of publication bias affecting their results by calculating the fail-safe ratio. They note that 14 studies would have to have gone unpublished for every published study in order for their meta-analytic results to be nonsignificant. While this ratio is intended to seem unreachably high, a couple of points should be kept in mind. First, the fail-safe ratio speaks not to unpublished studies but rather to unpublished
analyses. As discussed above, possibilities for alternative analyses (i.e., indirect cG×E replications) abound: alternative outcome, genotypic, and environmental variables can be investigated; covariates or additional moderators can be added to the model; additive, recessive, and dominant genetic models can be tested; phenotypic and environmental variables can be transformed; and the original finding can be tested in subsamples of the data. We observed each of these situations at least once among studies “consistent with” or “replicating” the original 5-HTTLPR-by-stressful life events interaction, and such indirect replications can have a high false positive rate. Second, and most importantly, Karg et al. used extremely liberal inclusion criteria, analyzing many indirect replications that we either classified as novel studies or excluded completely. Thus, the findings of Karg et al. and the findings we present here recapitulate one another; almost all novel studies (our review) and indirect replications (the Karg et al. meta-analysis) are positive, whereas most direct replications are not. This suggests that positive meta-analytic findings become more likely as study heterogeneity increases. Notably, this is exactly the opposite of what would be expected if the original results were true. Stricter replication attempts should be more likely, not less likely, to be significant. Rather than interpreting the fail-safe ratio as evidence that the 5-HTTLPR-by-stressful life events interaction has replicated, this ratio might be better interpreted as providing a rough estimate of how large the “file drawer problem” is in the cG×E field.
Conclusion
Despite numerous positive reports of cG×Es in the psychiatric genetics literature, our findings underscore several concerns that have been raised about the cG×E field in psychiatry. Our results suggest the existence of a strong publication bias toward positive findings that makes cG×E findings appear more robust than they actually are. Almost all novel results are positive, compared with less than one-third of replication attempts. More troubling is evidence suggesting that replication studies, generally considered the sine qua non of scientific progress, are also biased toward positive results. Furthermore, it appears that sample sizes for null replication results must be approximately six times larger than sample sizes for positive replication results in order to be deemed publishable on their own. Such a publication bias among replication attempts suggests that meta-analyses, which collapse across replication results for a given cG×E hypothesis, will also be biased toward being unrealistically positive. Although methods exist to detect publication biases (e.g., the funnel plot), they are not very sensitive, and correcting meta-analytic results for this bias is difficult (
41). Finally, our findings suggest that meta-analyses using very liberal inclusion thresholds (e.g., Karg et al. [
39]) are virtually guaranteed to find positive results.
The statistical power to detect cG×E effects is another important consideration. Unless cG×E effects are many times larger than typical genetic main effects, most cG×E studies conducted to date have been underpowered. This has several implications. The most obvious is that true G×E effects may often go undetected. However, low power also increases the rate of false discoveries across a field. Given the potentially low prior probability of true cG×E hypotheses, stemming from the difficulty of identifying the correct genetic and environmental variables, the false discovery rate in cG×E research in psychiatry could be very high; the possibility that most or even all positive cG×E findings in psychiatry discovered to date represent type I errors cannot be discounted.
For scientific progress to be made in the cG×E field, it is crucial to begin to differentiate the true cG×E effects from the false. How can this be accomplished? One step forward would be to encourage authors to submit and editors to accept null reports in order to reduce the publication biases present in the field, but incentives to publish positive reports are unlikely to change for either authors or editors anytime soon. Perhaps a more realistic way to begin discerning true results in the cG×E field is to acknowledge that false positive results are a natural consequence of the incentive structure that exists in modern science, and that because of this, authors, consumers, editors, and reviewers should recalibrate their views on what constitutes an important scientific contribution. Given the likely high false positive rate among novel findings (
19) and indirect replications (
36,
37) and the low false positive rate among direct replications (
36), well-powered studies conducted with the express purpose of closely replicating previous findings should be viewed as more scientifically important than novel “groundbreaking” cG×E results or indirect replications. The practice of according the most prestige to novel findings contributes to the ambiguous state of cG×E research and potentially to the proliferation of type I errors.
This review should not be taken as a call for skepticism about the G×E field in psychiatry. We believe that G×Es are likely to be common and that they may well prove to be important or even central for understanding the etiology of psychiatric disorders. At issue is how to separate the wheat from the chaff: which G×E findings are replicable and illuminating, and which are spurious and lead to wasted resources, false hope, and increased skepticism? Scientists investigating genetic main effects using genome-wide association methods have made minimizing false discoveries a central creed of their enterprise (
10). Indeed, the benefits of comprehensive SNP coverage and a conservative alpha have yielded hundreds of robust and replicable genetic associations. Such genome-wide methods have been proposed for the study of G×Es (
42) and will undoubtedly prove informative, but this is not the only solution. Rather, true progress in understanding G×Es in psychiatry requires investigators, reviewers, and editors to agree on standards that will increase certainty in reported results. By doing so, the second decade of G×E research in psychiatry can live up to the promises made by the first.
Acknowledgments
The authors thank Gary McClelland for his help with statistical concepts and John Hewitt, Soo Rhee, and Matthew McQueen for comments and suggestions on the manuscript.