The molecular and cellular underpinnings of psychosis in diseases like schizophrenia are unknown yet remain essential knowledge for rational treatment development. The hippocampus has been implicated in schizophrenia, and the repeated demonstration of changes in its structure and function have grown convincing. Studies show reduced hippocampal volume (
1), abnormal in vivo function (
2–
4), and replicable molecular pathology (
5) reliably across laboratories. Declarative memory, known to depend on the conjunctive memory function of the hippocampus, is one of the most consistently impaired cognitive functions in schizophrenia (
6–
9). In vivo biomarkers of hippocampal dysfunction in schizophrenia characteristically correlate with the psychotic symptomatology in individuals who are medication free (
10,
11).
The study of hippocampal subfield function in schizophrenia has already proven generative (
12). The subfields themselves (the dentate gyrus, the cornu ammonis [CA3, CA2, CA1], and the subiculum) have distinct and sequential functions in declarative memory formation (
13) and are differentially affected in the illness (
14). Excitatory projections connecting subfields have a low firing threshold, creating a unique hippocampal capacity for plasticity that advantages learning and memory (
15) but that, under pathological circumstances, such as in psychosis, can be a liability. CA3 contains an extensive network of recurrent collateral connections that represent the anatomic substrate of conjunctive encoding and pattern completion processes and that create the basis for declarative memory performance (
16) as well as an opportunity for pathological hyperassociation, as postulated in psychosis. In contrast, CA1 receives its strongest afferent stimulus from CA3 and shows a slower plasticity than CA3 for stabilizing place coding, while tuning multiple inputs dynamically from CA3 and the entorhinal cortex (
17).
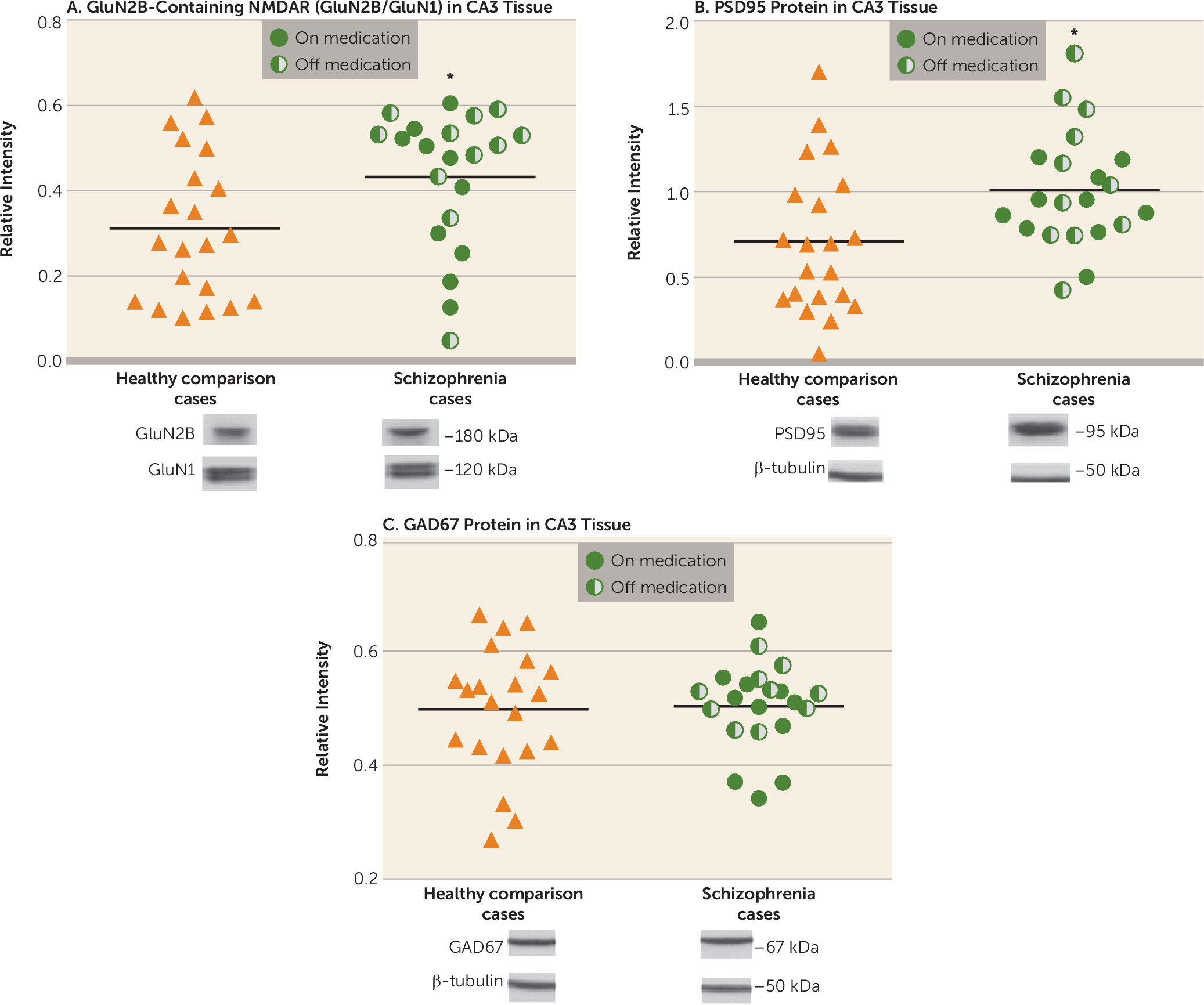
In this study, we examined CA3 tissue pathology in schizophrenia, contrasting the molecular changes in CA3 with those in CA1, postulating an increase in the specific molecular and cellular biomarkers of activity-dependent signaling in CA3. We previously articulated (
18) a model of psychosis in schizophrenia based on evidence of increased neuronal excitability in the hippocampus and of a reduction in afferent stimulation to CA3 from the dentate gyrus, a state we postulated to be mediated by increased long-term potentiation in CA3 and to result in neuronal hyperactivity downstream. The characteristic molecular determinants of increased long-term potentiation are well described in basic laboratory studies as being increases in “immature” GluN2B-containing NMDA receptors and in PSD95 or SAP102, both accompanied by synaptic remodeling (increased spine number), representing synaptic strengthening (
18). Specifically, based on this hippocampal psychosis model and testing the hypothesis of increased long-term potentiation in CA3, we postulated that GluN2B-containing NMDA receptors would be increased in CA3 in schizophrenia, with increased PSD95 protein, representing new synapses, along with anatomic evidence of spine proliferation, but that these would not be present in CA1, even though the increased neuronal activity generated in CA3 would be transmitted downstream to CA1. This hypothesis, if supported, would suggest increased long-term potentiation in CA3, supported molecularly and anatomically, that could plausibly be associated with hippocampal hyperactivity, mistakes of memory, and false memories with psychotic content in schizophrenia. These observations would support the concept of psychosis as a pathological alteration of hippocampal neuroplasticity resulting in alterations of normal learning and memory processes (
19,
20).
Discussion
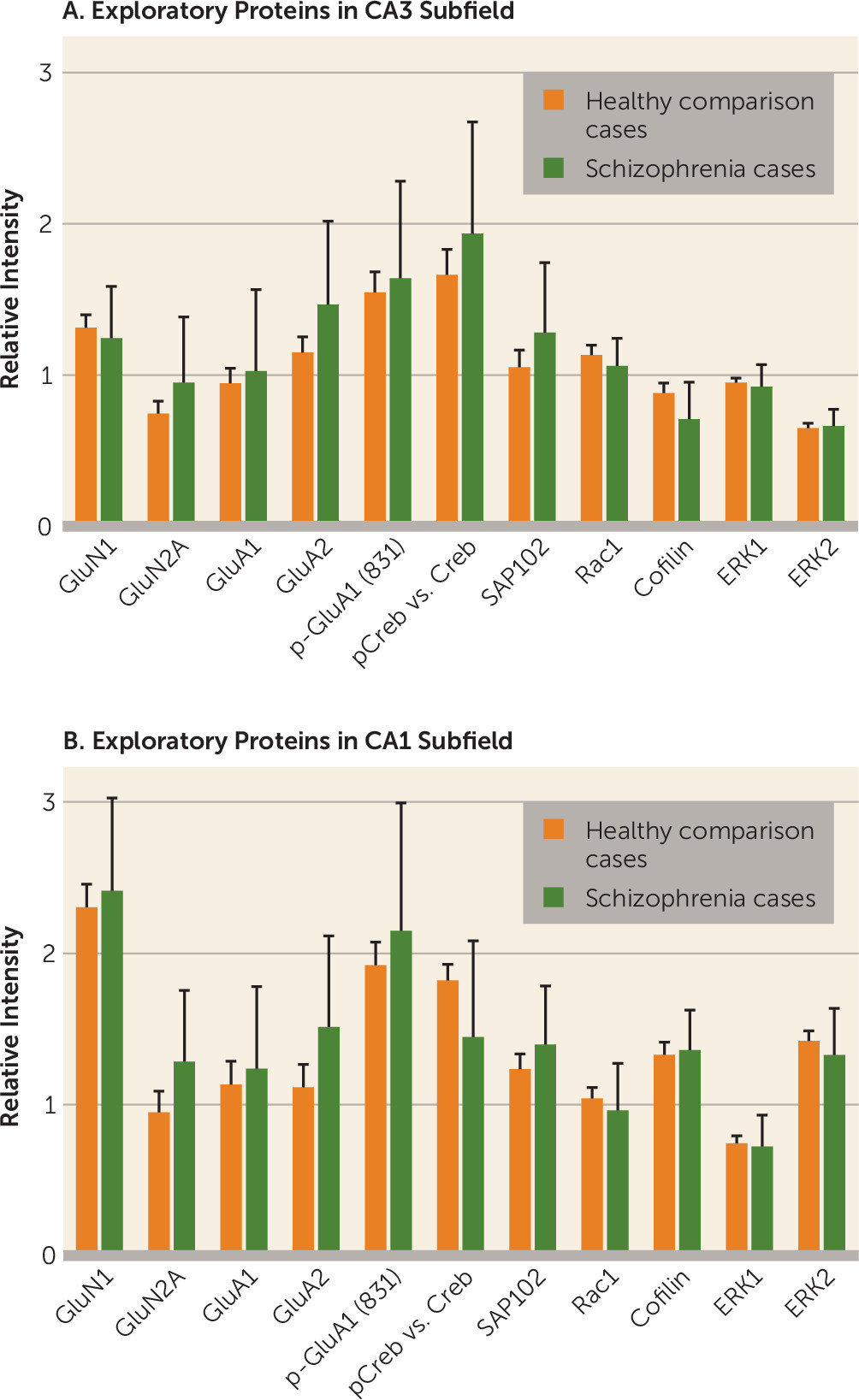
In this study, we contrasted tissue from healthy and schizophrenia cases on molecular and anatomic markers of synaptic plasticity within the CA3 hippocampal subfield, and produced evidence of altered plasticity in CA3 characterized by an increase in GluN2B-containing NMDA receptors and PSD95 protein and an increase in spine density. We did not observe changes in these cellular and molecular markers in CA1. The increases in GluN2B-containing receptors, the early developmental variant of the NMDA receptor, and in PSD95 protein occur without indication of AMPA receptor subunit alterations. The GluN2B/GluN1 increase in CA3 was present in the entire schizophrenia cohort, without any difference between cases on and off antipsychotic medication, indicating that this is a disease effect and not a chronic medication effect. These data are consistent with our previous report of increased BDNF mRNA in CA3 in schizophrenia, which also points to an increase in excitatory signaling in CA3 (
20). The mossy fiber innervations in CA3 contact both the excitatory pyramidal neurons at thorny excrescences and inhibitory interneurons by way of
en passant synapses, and these innervations have inverse effects on CA3 excitation (
25). Thus, strengthened transmission at the pyramidal cell and reduced inhibitory control of local interneurons occur together, augmenting CA3 pyramidal cell excitation from two sites, advantaging feed-forward excitation (
26). Uncontrolled feed-forward excitation may be the cerebral process that fuels hyperassociation, false memories, and psychotic mental events.
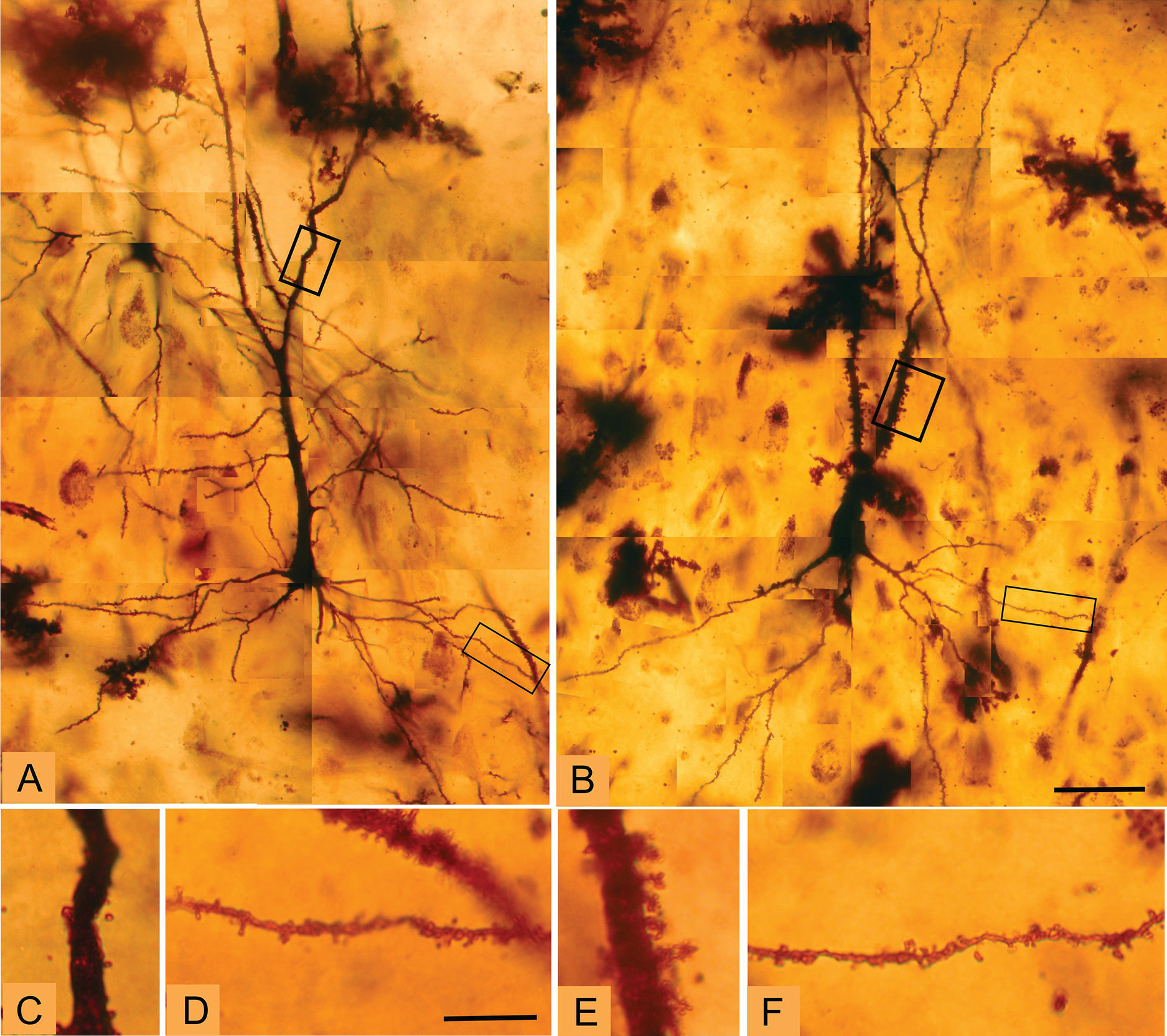
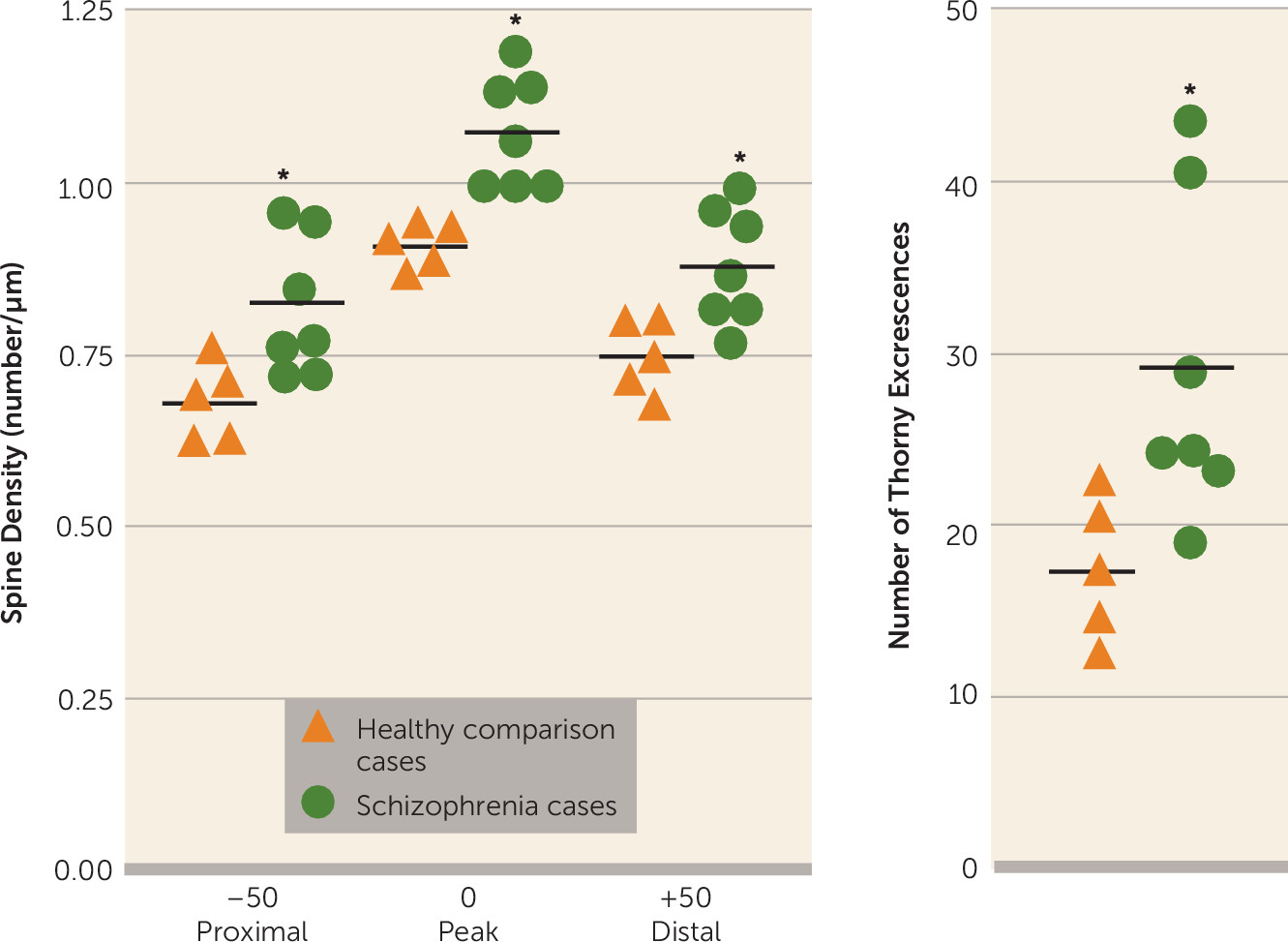
Golgi staining was used to examine the morphologic correlates of these molecular changes in pyramidal cell architecture in schizophrenia. Two distinct cellular regions of the CA3 pyramidal dendritic tree were examined: the proximal apical and basal dendrites corresponding to the CA3 stratum radiatum and the CA3 stratum oriens substrata, respectively. The anatomic changes show a clear increase in spine density limited to the stratum radiatum, at the apical trunk of pyramidal CA3 neurons, but not in the stratum oriens at the insertions of the recurrent collaterals. An increase was detected in the number of mossy fiber receptive sites, the thorny excrescences, in the stratum radiatum. The presence of greater spine density in the stratum radiatum is consistent with and could represent the morphologic manifestation of increased GluN2B-containing NMDA receptors in CA3 in schizophrenia, particularly as the GluN2B subunit advantages long-term potentiation (
27,
28). Increased spine density in CA3 is also compatible with the molecular findings of elevated PSD95, as overexpression of this protein has been shown to elevate spine density in hippocampal cultures (
29). Increased spine number is regularly observed following long-term potentiation-mediated increases in synaptic strength at excitatory synapses (
30,
31). These changes are compatible with evidence from the electron microscopy study of Kolomeets et al. (
32) showing that the specific number of mossy fiber/CA3 synapses is reduced, an effect that would lead to an increase in excitability and synaptic strength in CA3 itself through metaplasticity mechanisms. The specificity of morphometric abnormalities to sublayer components of the dendritic tree is intriguing and suggests that there may be a complex pattern of pathology in hippocampal subfields and their sublayers that could be related to afferent input patterning. Future studies may target morphometric features of the distal apical dendrites, which receive perforant path afferent input from the entorhinal cortex to further address sublayer specificity as well as extend the morphologic studies to substrata of the CA1 region.
These outcomes, including the increase in spines, an increase in PSD95, and the increase in GluN2B-containing NMDA receptors in CA3, are consistent with alterations in excitatory signaling in the hippocampus in CA3. This could represent an increase in neuronal excitability and long-term potentiation within CA3; moreover, we have previously suggested (
18) this as a model of altered metaplasticity dynamics in CA3, based on reduced afferent stimulation from the dentate gyrus, elevated sensitivity, and increased direct stimulation from the entorhinal cortex. Alternatively, or in addition, there could be an increase in silent synapses in CA3, which would prime the synapse for an increase in experience-dependent plasticity with synaptic “unsilencing.” Silent synapses are identified in laboratory preparations as synapses that exhibit NMDA-receptor-mediated electrical responses but no evidence of AMPA-receptor-mediated transmission; silent synapses are important during development (
33–
35). Recent studies have identified the generation of silent synapses in the mature brain during salient in vivo experiences (
36). It is thought that strong in vivo experiences—which, we suggest, might include acute psychotic experiences in schizophrenia—could selectively sensitize a neural circuit by generating silent synapses as a basis for robust increases in synaptic plasticity with relevant subsequent experience (
36). This interpretation is consistent with the presentation of psychosis, as most individuals with schizophrenic psychosis retain a propensity to psychosis after an initial psychotic episode and many show a cyclic recurrence of psychotic manifestations. The alteration in CA3 plasticity conditions with an initial psychotic episode could create a vulnerability to new excitatory input within CA3 after a florid psychosis, which could generate “runaway” activity within the recurrent collateral projections that would diminish prediction error mechanisms, increase associations, and allow false memory formation with psychotic content. Both of these interpretations are consistent with emerging schizophrenia genetics, which converge not only on strong risk genes like NRG1 (which shows an altered NRG1 fragment in hippocampus [
37]), DISC1, and CHRNA7 (
38), but also on functional gene networks, including those modifying glutamate, synaptic function, and neuronal plasticity. The 15q13-14 locus, which is implicated in schizophrenia, includes the alpha-7 nicotinic receptor promoter (
38); because cholinergic innervation is differentially directed to CA3 within the human hippocampus (
39) and largely localized to CA3 interneurons (
40), a loss of cholinergic capacity and subsequent failure of interneuronal inhibition could support CA3 hyperactivity. Also, a genetically manipulated mouse based on the 22q11.2 schizophrenia risk deletion shows an increase in hippocampal high-frequency synaptic transmission, increased long-term plasticity, and exaggerated dendritic spines (
41), suggesting these as disease elements in schizophrenia.
The CA3 subfield changes in schizophrenia tissue described here may provide the molecular and cellular substrate supporting hippocampal hyperactivity in vivo that has been well described in schizophrenia patients—that is, hyperactivity represented by increased hippocampal cerebral blood flow (
3,
42) and blood volume (
12). These molecular alterations were not seen in CA1, suggesting that the in vivo hippocampal hyperactivity that has been reported in CA1 is propagated downstream from CA3 (
12). The dynamic characteristics of NMDA receptor membrane insertion, removal, and translocation within neural synapses allow for their involvement in alterations of synaptic strength (
43,
44), recognizing that the regulation of synaptic strength at the mossy fiber-CA3 pyramidal cell synapse is unique and complex (
26,
33). This change could contribute to psychosis manifestations through pathological “pattern separation” (
45) and/or “pattern completion” computations (
46–
48).
In this study, we examined NMDA receptor subunits only in hippocampal subfields. A large literature exists on these subunits in other schizophrenia-affected brain regions (
49) and in whole hippocampus (
50), prefrontal cortex (
51), thalamus (
52), cerebellum (
53), and substantia nigra (
54). Changes, especially in the prefrontal cortex, have been variable across studies (
55–
57). The relationship between the molecular changes reported here in hippocampal subfields and the neocortical anatomic and molecular neuropathology of schizophrenia reported elsewhere (
58,
59) is not clear. These changes could be independent, correlated, or interdependent, a question important for future investigation. The coexistence of both hippocampal hyperactivity (plausibly associated with psychosis) and prefrontal cortical hypoactivity (plausibly associated with cognitive impairment) could be functionally linked if the hyperexcitatory efferents from the hippocampus to prefrontal cortical targets fell primarily onto the affected populations of cortical inhibitory interneurons, contributing to the dysregulation of GABA inhibitory control of prefrontal pyramidal neurons.
A classic hypothesis for hippocampal hyperactivity in schizophrenia has been disinhibition of hippocampal pyramidal cells by a reduction in the inhibitory GABA-mediated modulation of pyramidal cell firing (
60). Cell-counting studies in the hippocampus contrasting schizophrenia and bipolar disorder patients with healthy subjects have reported metabolic (
61) and molecular (
2) alterations in bipolar disorder, largely sparing the hippocampus in schizophrenia, although previous studies found inhibitory deficits in the hippocampus in schizophrenia (
62,
63). Yet, in our study, in neither CA3 nor CA1 tissue from schizophrenia cases did we detect reductions in GAD67 protein. Further research is clearly needed to examine subtle effects of changes in inhibitory GABA modulation in CA3.
In summary, these data show changes in schizophrenia in the GluN2B-containing NMDA receptor in CA3 of the hippocampus and not in CA1, along with consistent increases in PSD95 and in pyramidal cell spine number. These findings are consistent with increased neuronal excitability in CA3 and/or with an increase in silent synapses in CA3 in schizophrenia, conditions that could plausibly underlie manifestations of psychosis in the illness. The findings have implications for potential molecular markers in the illness, for novel medication targeting with further confirmation, and for use in modeling schizophrenia psychosis in animal paradigms.