The pathophysiology of OCD and tic disorders more broadly is incompletely understood (
28,
29). Substantial evidence implicates dysregulation of cortico–basal ganglia circuits in both conditions (
30–
35), as well as in PANDAS. The striatum, which in primates consists of the caudate and putamen, is the primary input nucleus of the basal ganglia and integrates synaptic inputs from limbic and cortical regions (
36). The vast majority of neurons in the striatum (∼95% in rodents) consists of GABAergic medium spiny neurons. Only a small fraction (∼5%) are interneurons (
37), of which GABAergic interneurons are the most abundant. They can be classified on the basis of their expression of markers such as parvalbumin (PV), somatostatin, neuropeptide Y, and neuronal nitric oxide synthase (nNOS). A distinct, small population of large, tonically active cholinergic interneurons (CINs) is characterized by expression of choline acetyltransferase (ChAT). Although few in number, these interneurons are key regulators of striatal function (
38–
41). Both CINs and PV-expressing GABAergic interneurons are reduced in number in the caudate and putamen of individuals with Tourette’s syndrome (
42–
44). Transcriptomic analysis of postmortem striatum from individuals with Tourette’s syndrome similarly reveals reduced expression of interneuron-associated genes (
43). Experimental depletion of these cells in developmentally normal mice produces repetitive behavioral pathology (
45,
46), suggesting that this neuronal deficit is causally related to symptomatology.
Methods
PANDAS Serum
These investigations of human serum samples were approved by the Human Investigations Committees of Yale University and the National Institute of Mental Health (NIMH). Parents gave written informed consent for participation in the study, and children gave assent. In the first two cohorts of patients analyzed, sera from 11 children with a well-substantiated diagnosis of PANDAS were selected from a clinical trial performed at NIMH and Yale University (ClinicalTrials.gov identifier, NCT01281969), as described previously (
21). Participants met rigorous clinical and laboratory criteria for a diagnosis of PANDAS (
5) and were additionally selected for having a robust clinical response to treatment with intravenous immunoglobulin (IVIG) (
21); we reasoned that IVIG responders are most likely to have antibody-mediated pathology. The Children’s Yale-Brown Obsessive Compulsive Scale (CY-BOCS) (
48) was administered at baseline (before IVIG treatment) and at 6, 12, and 18 weeks; serum from the baseline and 12-week visits (visits 1 and 3 in the clinical trial; see Figure S1 in the
online supplement) were used in the present study. The mean CY-BOCS score was 28.64 (SD=3.41) at baseline and 5.64 (SD=5.26) at 3 months, after one or two rounds of IVIG treatment. Samples from a third cohort of 16 PANDAS patients and 12 matched control subjects were obtained from the same NIMH PANDAS clinic; these samples were drawn from several different studies and were not preselected for IVIG response. Samples from age- and gender-matched healthy subjects with no clinically significant OCD or tic symptoms were collected by the same investigators at NIMH and were stored and tested in parallel. The first patient cohort, consisting of five PANDAS patients and five matched control subjects, has been described previously (
47). All samples were aliquoted into small volumes on arrival at Yale University and stored at −80°C until use. Demographic data for all participants are summarized in Tables S1 and S2 in the
online supplement. All sera were coded before being sent from NIMH to Yale University for analysis, and all analyses were performed blind to diagnosis and treatment condition.
Normal Human Brain Tissue
The collection and preparation of formalin-fixed normal human brain tissue have been described previously (
43). Coronal sections of caudate and putamen (50 µm, two sections per serum) were used for serum antibody binding and immunostaining, as further detailed below.
Animals
All experimental procedures were approved by the Yale University Institutional Animal Care and Use Committee, in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Male and female C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, Me.). Double-transgenic D1-DARPP-32-FLAG/ D2-DARPP-32-Myc were backcrossed to C57BL/6J for at least nine generations and have been described previously; the transgenically expressed FLAG and Myc epitope tags allow dissociable immunostaining of D1R- and D2R-expressing medium spiny neuron populations (
49,
50). All mice were maintained on a 12-hour light/dark cycle and used at 3–6 months of age.
Reagents and Antibodies
N-methyl-d-glucamine (NMDG), glucose, thiourea, sodium pyruvate, sodium ascorbate, tetrodotoxin (TTX), Sudan Black B, and Triton X-100 were obtained from Sigma (St. Louis, Mo.). Dopamine and other chemicals used for electrophysiology (see Figure S12 in the online supplement) were also obtained from Sigma, with the exception of 5-HT and AMPA, which were obtained from Tocris (Ellisville, Mo.). Paraformaldehyde, KCl, and other chemicals used for acute brain slice analysis were obtained from J.T. Baker Chemical Co. (Phillipsburg, N.J.). Ketamine was obtained from Zoetis (Madison, N.J.). Xylazine was obtained from Akorn, Inc. (Decatur, Ill.). Antibodies used in immunohistochemical staining are listed in Table S3 in the online supplement.
Determination of Serum IgG Titer and IgG Depletion
Total IgG titers in control and PANDAS sera were determined using the IgG (Total) Human ELISA Kit (Thermo Scientific, Rockland, Ill.), following the manufacturer’s instructions. Prediluted sera were added to microplates precoated with an anti–human IgG capture antibody and incubated for 2 hours at room temperature. After washes, a horseradish peroxidase (HRP)–conjugated anti–human IgG detection antibody was added for 1 hour at room temperature. After extensive washes, 3,3′,5,5′-tetramethylbenzidine solution was added to each well for 15 minutes. The reaction was stopped with 1 M phosphoric acid and optical density reading was obtained using an MQX200 μQuant plate reader (BioTek, Winooski, Vt.) at 450 nm. Human IgG standard provided in the kit was used as a reference to determine IgG concentration in each serum. All sera were diluted in 1× phosphate-buffered saline (PBS) + 0.1% bovine serum albumin to 500 mg/dL IgG.
For IgG depletion, baseline (pre-IVIG treatment) PANDAS sera from 11 patients (see Table S1 in the online supplement) were incubated with Protein A/G agarose beads (Thermo Scientific) for 2 hours at 4°C. Supernatants were collected by brief centrifugation (1000×g for 1 minute). Supernatants were mixed with fresh Protein A/G agarose beads for a second round of depletion. To confirm IgG depletion, small aliquots from both batches of supernatants were tested on dot blot using a rabbit anti–human IgG antibody (Abcam, Cambridge, Mass.) and HRP-conjugated goat anti-rabbit secondary antibody (Thermo Scientific). Membranes were developed using the Chemiluminescent Substrate kit (Thermo Scientific) and visualized on a ChemiDoc XRS+ system (Bio-Rad, Hercules, Calif.).
Immunohistochemistry
Mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) with xylazine (10 mg/kg) and transcardially perfused with cold 4% paraformaldehyde in 1×PBS (pH 7.4). Brains were fixed overnight in 4% paraformaldehyde at 4°C, followed by equilibration in 30% sucrose for 48 hours at 4°C. Striatal slices were cut at 20 μm using a Leica CM3050S cryostat (Leica, Buffalo Grove, Ill.). Slices were stored in a cryoprotectant solution (30% glycerin, 30% ethylene glycol in 1×PBS, pH 7.4) at −20°C until use. All staining was done using wild-type (WT) C57BL6 mice, except for examination of D1R- and D2R-expressing medium spiny neurons, for which we used D1-FLAG/D2-Myc double-transgenic mice (
49,
50).
Brain sections were washed three times, 10 minutes each time (3×10 minutes), in 1×PBS (pH 7.4) to remove cryoprotectant, followed by incubation in freshly prepared 0.1% Sudan Black (in 70% ethanol) for 10 minutes at room temperature to reduce autofluorescence. Slices were washed 3×10 minutes in 70% ethanol and 3×10 minutes in 1×PBS. Slices were blocked in 1×PBS+0.3% Triton X-100 supplemented with 5% donkey serum (Jackson ImmunoResearch, West Grove, Pa.) for 1 hour at room temperature and then incubated overnight at 4°C with PANDAS or healthy control serum at 1.25 mg/dL (diluted in blocking buffer; this dilution was found in extensive pilot experiments to provide an optimal signal-to-noise ratio for cell body staining). For staining with mouse primary antibodies (anti-PV, anti-nNOS, and anti-FLAG), slices were additionally blocked in Mouse-on-Mouse reagent (Vector Laboratories, Burlingame, Calif.), following the manufacturer’s instructions. Slices were then washed 3×10 minutes in 1×PBS+0.3% Triton X-100 and incubated with primary antibodies (see Table S3 in the online supplement) in blocking buffer overnight at 4°C. Sections were double-immunostained with anti–human IgG to identify serum IgG binding (green) and selected neuronal markers (ChAT, PV, nNOS, and antibodies to the epitope tags FLAG and Myc to detect medium spiny neurons in D1-DARPP-32-FLAG/D2-DARPP-32-Myc; red). The next day, slices were washed 3×10 minutes in 1×PBS+0.3% Triton X-100 and then incubated with fluorophore-conjugated secondary antibodies (see Table S3) for 1 hour at room temperature. After 3×10 minute washes in 1×PBS, slices were mounted in Vectashield HardSet Mounting Medium (Vector Laboratories), coverslipped, and stored at 4°C.
For illustrative purposes, IgG binding was visualized by sequential scanning of slices on an Olympus Fluoview FV-1000 confocal microscope with a 40×/1.30 NA objective (Olympus, Japan; Figures 1A, 3A, 3F, and 4A, and corresponding high-magnification panels in the supplementary figures).
For quantification, we used lower-magnification images collected using single-photon fluorescence imaging; these images are of lower resolution than confocal images and do not resolve the z-dimension as well, but they allow us to quantify an order of magnitude more cells (see Figures S2 and S16–S19 in the online supplement). Single-photon fluorescence images were collected using an Axioskop 2 fluorescent microscope with a 20×/0.8 NA objective (Zeiss, Oberkochen, Germany) or an Axio Scope A1 fluorescent microscope with a 10×/0.45 NA objective (Zeiss). We took 36–48 images from four to six mice per serum in the dorsal striatum and 12–16 images from four to six mice for each serum in the medial septum. Microscope settings were kept constant (2500−3500 ms exposure for IgG images and 1000–1500 ms for neuronal markers: ChAT, PV, nNOS, FLAG, and Myc) across all images between which comparisons were made. In some cases (Figure 1; see also Figure S6 in the online supplement), because of limited microscope availability, images were captured using different microscopes for different cohorts. To compensate for differences in background binding across experiments, data were normalized to the mean value of the control samples processed in the same batch. Microscope fields were chosen without overlap within dorsal striatum, using the lateral ventricle as an anatomical landmark, to ensure that comparable fields were examined for all sera; the distribution of images and the total number of cells of each type quantified are shown in Figure S2 in the online supplement. Cell counts did not differ between PANDAS and control groups for any neuron type.
Human brain sections were triple-immunostained with anti–human IgG and markers for ChAT and PV, following the same protocol. Three-color images were collected using an Olympus Fluoview FV-1000 confocal microscope with a 40×/1.30 NA objective. Triple-staining and confocal imaging limit the number of cells quantified but were used for this experiment to permit triple-labeling and thereby conserve human tissue samples.
Acute Brain Slice Preparation, Ex Vivo Serum Treatment, and Quantification of Phospho-rpS6
For the functional assay shown in Figures 3 and 5, acute brain slices were prepared from male and female wild-type mice and treated ex vivo with PANDAS serum (pre-IVIG, post-IVIG, and IgG-depleted pre-IVIG) or control serum. Mice were sacrificed by cervical dislocation; brains were quickly removed and placed in ice-cold oxygenated NMDG-based artificial cerebrospinal fluid (aCSF), as previously described (
51). We cut 100 μm coronal slices through the striatum (see Figure S2 in the
online supplement) using a Leica VT1000S vibratome (Leica Microsystems, Bannockburn, Ill.) in the NMDG solution containing (in mM) 92 NMDG, 2.5 KCl, 1.25 NaH
2PO
4, 30 NaHCO
3, 20 HEPES, 10 MgSO
4, 0.5 CaCl
2, 25 glucose, 2 thiourea, 3 sodium pyruvate, and 5 sodium ascorbate, pH 7.35, saturated with 95% O
2/5% CO
2.
Slices were recovered in NMDG-aCSF for 10 minutes at 32°C before being transferred to regular aCSF containing (in mM) 119 NaCl, 2.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 2 CaCl2, 2 MgSO4, and 12.5 glucose for 1 hour at 30°C under constant oxygenation with 95% O2/5% CO2. After recovery, slices were treated with control drugs (TTX 1 μM; KCl 20 mM; 0.1% dimethyl sulfoxide (DMSO) in aCSF), control serum (1.25 or 6.25 mg/dL), or PANDAS serum (baseline, post-IVIG, or IgG-depleted baseline; 1.25 or 6.25 mg/dL) for 1 hour at 30°C. A subset of slices were treated with aCSF with 0.1% DMSO in each experiment; data from other conditions were normalized to this vehicle condition for analysis. After treatment, slices were fixed in cold 4% paraformaldehyde in 1×PBS (pH 7.4) containing 5 mM NaF for 1 hour at 4°C.
Sections shown in Figure 3 were double-immunostained for phospho-rpS6 (green) and ChAT (red) or PV (red), as described above. For ChAT/P-rpS6 staining, 23–56 images from four to seven mice were taken in the dorsal striatum for each serum and 100–350 CINs were quantified for each serum. For PV/P-rpS6 staining, 40–52 images from five mice were taken in the dorsal striatum and 60–100 PV+ interneurons were quantified for each serum. Total cells counted did not differ between PANDAS and control groups. Sections shown in Figure 5 were triple-immunostained with anti–human IgG, anti-ChAT, and anti-PV, as described above. Images were collected using an Olympus Fluoview FV-1000 confocal microscope with a 40×/1.30 NA objective. Triple-immunostaining and confocal imaging limit the number of cells quantified but were used for this experiment to conserve serum.
Image Processing and Quantification
Automated quantitation of mean fluorescence intensity within each cell was achieved using Fiji ImageJ from NIH (
https://imagej.net/Fiji/Downloads) with batch processing, as illustrated in Figure S3 in the
online supplement. Neuronal marker immunostaining (ChAT, PV,
N-NOS, FLAG, or c-Myc) was thresholded and used to generate regions of interest corresponding to cell bodies of the selected cell type. The number and spatial extent of these cellular regions of interest were quantified to ensure between-group comparability. The total regions of interest per section obtained for each serum was interpreted as cell number for the selected cell type. This spatial filter was then overlaid on the corresponding anti–human IgG or phospho-rpS6 image, and immunostaining within each cellular region of interest was quantified. Background IgG or phospho-rpS6 signal was subtracted to generate an adjusted value for each cell. This procedure is illustrated in Figure S3. Although this procedure is wholly automated, serum identity was coded until all the values were retrieved, as an additional safeguard for the rigor of the analysis.
Brain Slice Electrophysiology
Brain slices were prepared as previously described (
52). Briefly, mice were anesthetized using chloral hydrate (400 mg/kg i.p.) and brains were removed and placed in ice-cold aCSF in which sucrose (252 mM) was substituted for NaCl (sucrose-aCSF). A block of tissue containing corticostriatal and coronal slices (400 μm) was cut in sucrose-aCSF with an oscillating-blade tissue slicer. After 60 minutes’ incubation with normal or PANDAS sera (6.25 mg/dL) in standard aCSF containing (in mM) 128 NaCl, 3 KCl, 2 CaCl
2, 2 MgSO
4, 24 NaHCO
3, 1.25 NaH
2PO
4, and 10
d-glucose (pH 7.35–7.38) equilibrated with 95% O
2/5% CO
2 at room temperature, slices were transferred into a submerged recording chamber; bath temperature was then raised to 32°C. Patch pipettes (3–5 MΩ) were pulled from glass tubing with a Flaming-Brown horizontal puller. The pipette solution contained the following (in mM): 115 K-gluconate, 5 KCl, 2 MgCl
2, 2 Mg-ATP, 2 Na
2-ATP, 10 Na
2-phosphocreatine, 0.4 Na
2-GTP, and 10 HEPES, pH 7.33. Neurobiotin (0.3%) was added to the pipette solution to mark cells for later imaging. All drugs, in aCSF, were applied for 1.5 minutes at a time in the fast-flowing bath, followed by a washout period of 8–10 minutes.
Cholinergic interneurons in striatum were visualized by videomicroscopy (40×IR lens) with infrared differential interference contrast and selected on the basis of their size and characteristic morphology. Whole-cell recordings were performed with an Axoclamp-2B amplifier. Postsynaptic currents were studied in the continuous single-electrode voltage-clamp mode (3000 Hz low-pass filter) clamped at −70 mV. Spontaneous or neurotransmitter (5-HT, AMPA, and dopamine) evoked spike and intrinsic membrane properties were determined using current clamp. Analysis of spike frequency and inward current was conducted with Clampfit, version 10 (Molecular Devices, Sunnyvale, Calif.).
Data Analysis
All data are expressed as means and standard deviations. Statistical analyses were performed using SPSS Statistics, version 24 (IBM, Armonk, N.Y.) or Prism, version 7.0 (GraphPad Software, La Jolla, Calif.). Significance (p<0.05) was determined by two-tailed t test (unpaired or paired), one-sample t test, or one-way or two-way analysis of variance with post hoc Tukey’s test. Specific tests used in each analysis are listed in the figure legends. The unit of analysis (patient/serum, mouse, slice, or cell) is indicated for individual analyses. Pearson correlation was used to examine linear relationships between measures. For electrophysiology, the mean discharge frequency at 30 seconds before administration and 30 seconds after administration was compared and statistically analyzed.
Discussion
An autoimmune pathophysiology has been proposed for PANDAS but remains unproven (
5–
7). While a number of possible pathogenic antibody targets have been described, none has been consistently replicated, and how the binding of antibodies to brain targets leads to symptoms remains unclear (
8,
9,
15–
18,
61–
63). Our findings, here and in our previously published pilot study (
47), reveal a hitherto unrecognized target for antibodies in these patients: the cholinergic interneurons (CINs) of the striatum. Our functional data (
Figures 3 and
5) suggest that these antibodies can bind to CINs and reduce their activity, thereby disrupting the normal function of the corticostriatal circuitry.
Disruption of CINs has previously been implicated in Tourette’s syndrome, and in repetitive behavioral pathology more generally. Specifically, CINs are reduced in number in the striatum of adults with refractory Tourette’s syndrome (
42,
43). Experimental depletion of these neurons in developmentally normal mice produces elevated grooming and repetitive behaviors, suggesting a causal relationship between this deficit and the development of relevant symptoms (
45,
46). These previous, independent results make CIN dysregulation an inherently plausible locus of pathology in PANDAS.
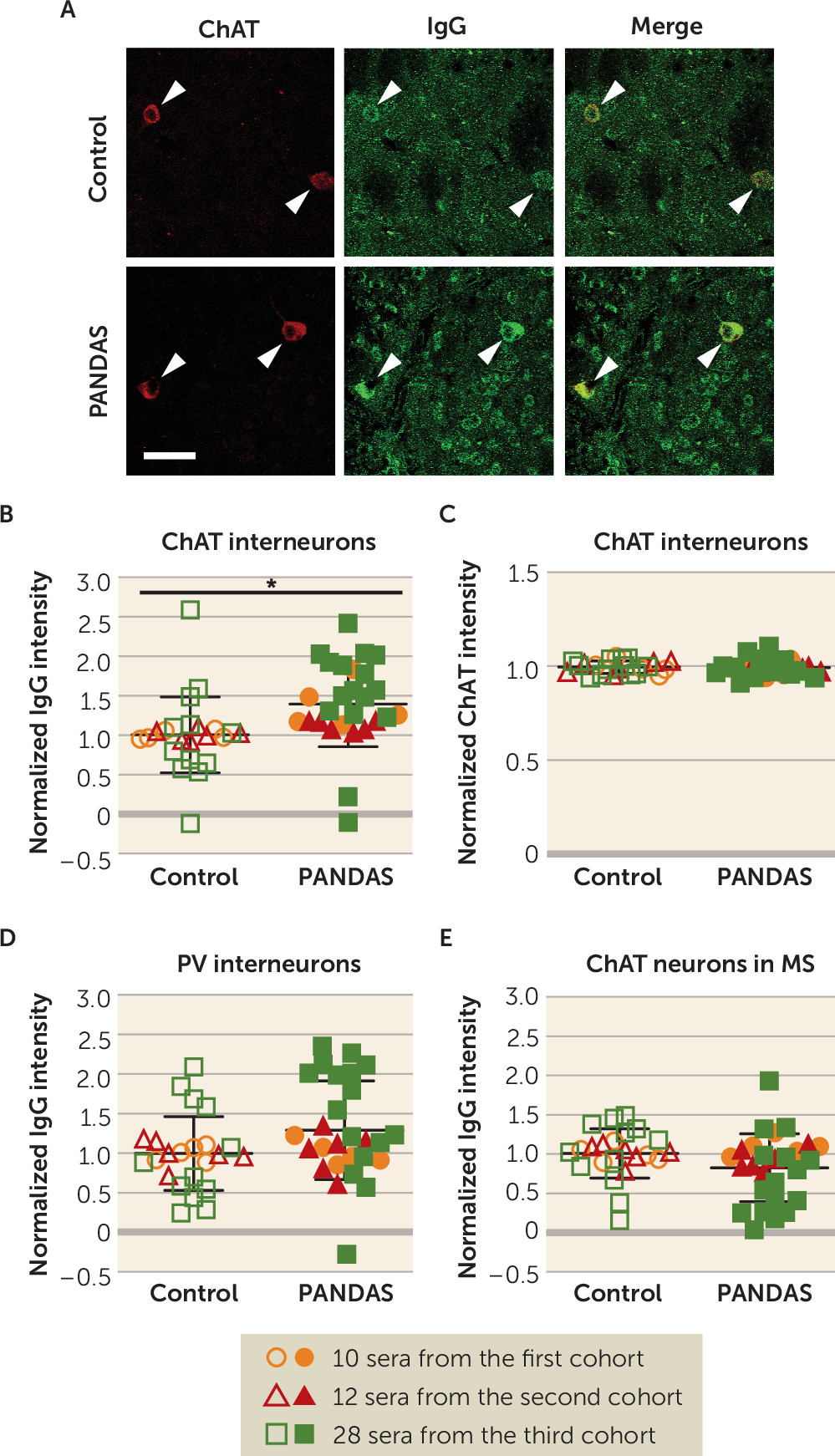
The consistency and specificity of elevated antibody binding to CINs, but not other examined cells, is striking. We found elevated IgG binding to mouse striatal CINs in 27 children with PANDAS, across three independent cohorts of patients, compared with age- and gender-matched control subjects (
Figure 1B). In conjunction with our previous results (
47), the effect has now been seen using two different experimental approaches (in vivo and ex vivo antibody delivery), with both confocal and standard fluorescent imaging, with two different quantification strategies, in three different primary experimenters’ hands, with blinding to experimental condition. There is, with equal consistency, no elevated binding to two classes of GABAergic interneuron in the striatum (
Figure 1D; see also Figure S6A in the
online supplement) (
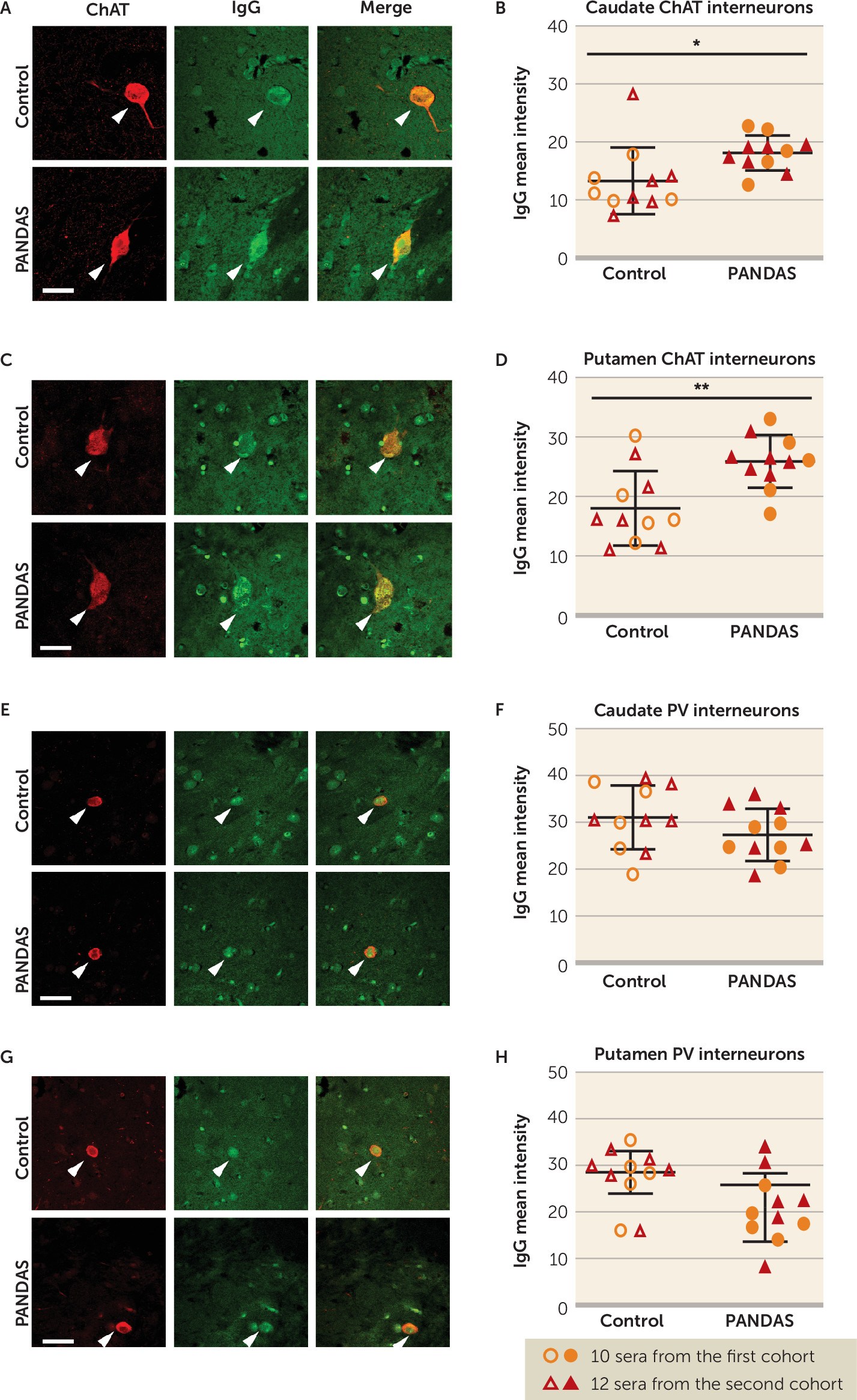
47). Moreover, we confirmed these findings in human brains: 11 PANDAS sera showed elevated IgG binding to CINs in both caudate and putamen, compared with matched control subjects (
Figure 2B,D), with no difference between groups in IgG binding to PV-interneurons (
Figure 2F,H). It is of course possible that differential binding to other cell types would be seen in a more exhaustive examination of different cell types throughout the brain; this is an important direction for future research.
We found no altered binding to D1R- or D2R-expressing medium spiny neurons of the striatum (see Figure S6D,G in the
online supplement). Binding by PANDAS antibodies to both D1R and D2R has been investigated previously, using in vitro binding to denatured protein (
17,
18) or to heterologous cells overexpressing dopamine receptors (
64). D2R is present on CINs and thus could explain elevated CIN binding. The lack of any change in binding to D2R-expressing medium spiny neurons, however, argues against this possibility. We also see no elevated binding to cholinergic cells in the medial septum (
Figure 1E), indicating that the abnormality does not generalize to cholinergic cells throughout the nervous system, or even the forebrain.
The specific molecular targets on striatal CINs that explain elevated binding by PANDAS IgG remain unclear. There may be a single molecular target on these cells that has eluded identification in previous studies, perhaps because it is present at low concentration in total striatal lysate (as CINs represent a small minority of the cells in the striatum). However, our experimental approach does not require that a single molecular target explain the observed binding in all patients; it is possible that there are multiple targets that can lead to similar binding patterns and similar pathophysiological effects in different patients. Such heterogeneity could further explain the lack of replication that has characterized efforts to identify specific antibodies on the basis of binding to specific molecules, rather than to cell types. We note that in these experiments (though not in our previous work [
47]) tissue was permeabilized before application of serum, and thus both cell surface and intracellular antigens are accessible; surface antigens may be more relevant pathophysiological targets in vivo. Identification of the molecular targets of antibody binding in individual sera is an important future direction for this work.
In the third cohort of PANDAS patients, who were not selected for IVIG response (and some of whom were not treated with IVIG at all), we found a similar but statistically weaker increase in IgG binding to CINs. The somewhat attenuated effect in this third cohort suggests that it may be more heterogeneous, containing some subjects with IgG-mediated pathology similar to that present in the first cohorts but others with a different pathophysiology. Identification of the clinical correlates of elevated IgG binding to CINs, both in patients with PANDAS and in related diagnostic groups, is an important direction for future work.
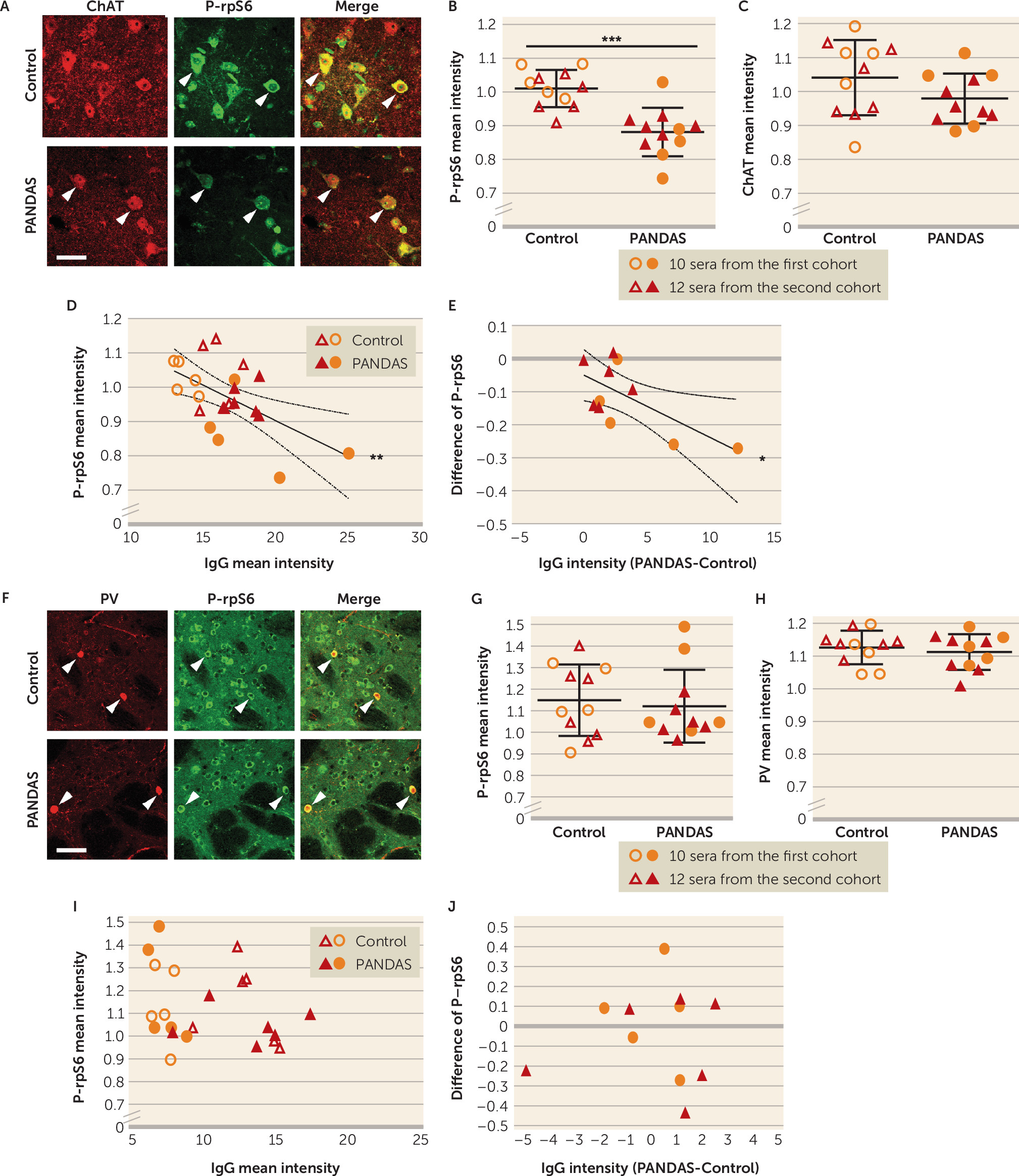
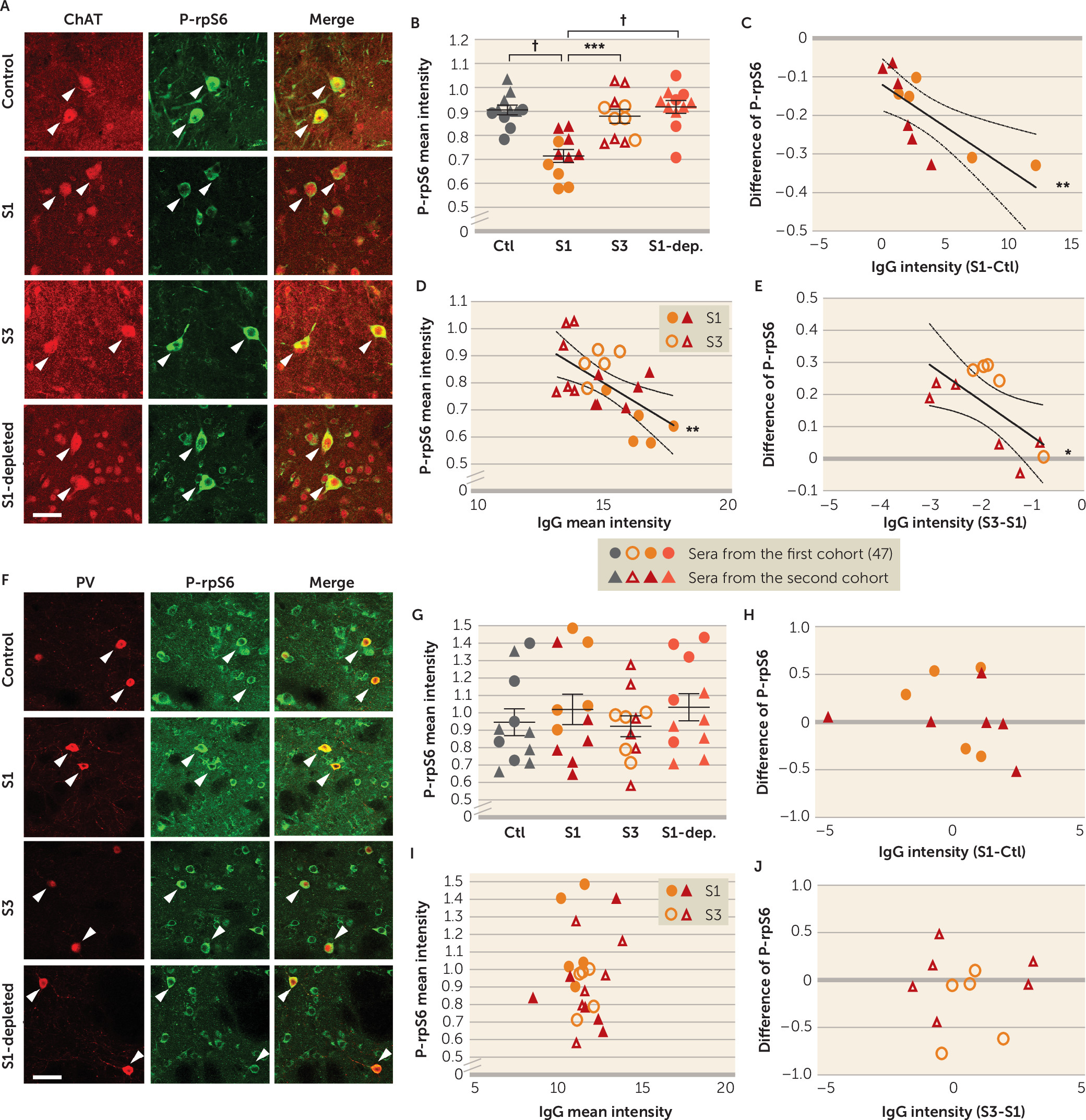
Our results also show, for the first time, that PANDAS antibodies can affect CIN activity, providing critical support for the proposed pathophysiological mechanism. Specifically, we treated mouse striatal slices with PANDAS and control serum ex vivo, and we measured activity-dependent phosphorylation of ribosomal protein S6 (P-rpS6), a validated marker of neuronal activity in both CINs (
57) and PV-expressing interneurons (
58,
59). We found reduced P-rpS6 after treatment with 11 PANDAS sera, but not with matched control sera (
Figure 3B). This effect is lost when IgG is depleted from PANDAS serum (
Figure 5B), indicating that IgG binding is necessary for CIN inhibition. Importantly, there is no similar effect on PV-expressing interneurons (
Figure 3G–J), further emphasizing the specificity of the effects on CINs seen throughout our study. Of course, we cannot exclude the possibility that antibodies in PANDAS sera can have other effects on PV-expressing interneurons not captured by this assay, or on other cells elsewhere in the brain.
Antibody-modulating therapies such as IVIG and plasmapheresis have shown promise in the treatment of PANDAS in some studies, though not in all (
19–
22). Our first two patient cohorts were drawn from a recent controlled study of IVIG treatment (
21) (see Figure S1 in the
online supplement). This allowed testing of serum collected before and after treatment (
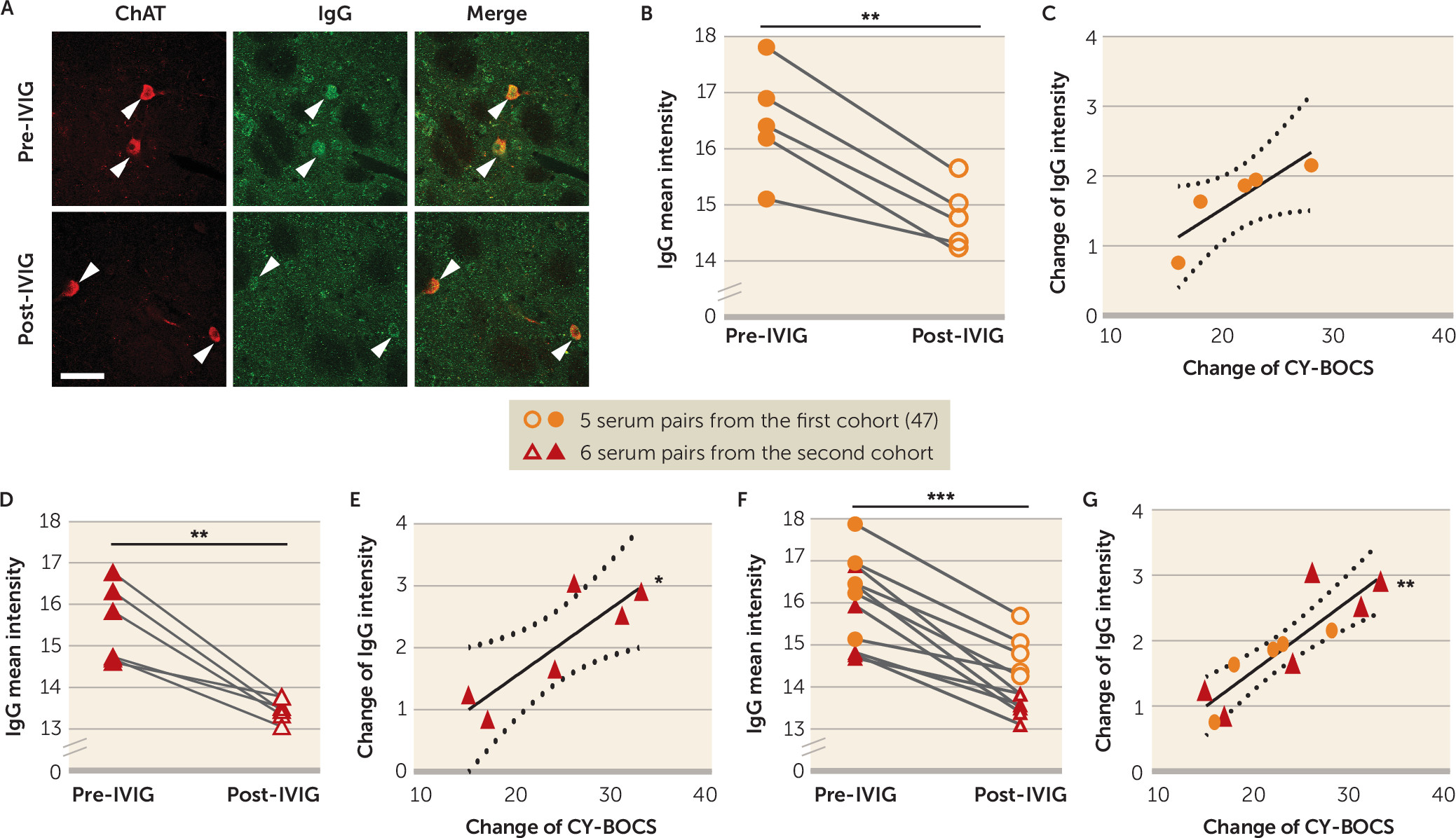
47). We found that IgG binding to CINs was reduced after IVIG treatment, and that this change correlated robustly with symptom improvement (
Figure 4). Similarly, post-IVIG serum did not reduce CIN activity (
Figure 5B); the change in antibody binding correlated with the change in ability to reduce CIN activity. Critically, there was no change in binding to PV-expressing interneurons in the striatum or in cholinergic cells of the medial septum after IVIG treatment (see Figure S14 in the
online supplement).
As an additional measure of neuronal activity, we examined CIN response to bath neurotransmitter treatment ex vivo after incubation with PANDAS or control serum. PANDAS serum significantly attenuated AMPA-induced spike frequency (see Figure S12A,B in the
online supplement). This may explain reduced P-rpS6 after PANDAS treatment (
Figures 3 and
5), to the extent that glutamate tone contributes to basal CIN firing in our ex vivo preparation. PANDAS serum also modulated cellular responses to both dopamine and 5-HT. We were only able to perform these electrophysiological experiments with a single serum pair, because of limited supplies of clinical serum and the relatively large amounts needed for these studies. Nevertheless, in conjunction with the P-rpS6 staining data across 11 PANDAS samples and matched control subjects (
Figure 3), these data indicate that PANDAS serum can reduce activity of CINs.
The downstream consequences of PANDAS IgG binding to CINs in vivo remain to be elucidated. We hypothesize that pathogenic PANDAS antibodies reduce CIN activity in vivo, as we have shown ex vivo (
Figures 3 and
5; see also Figures S12 and S13 in the
online supplement). This may functionally parallel the effect of reduced CIN number in adults with severe Tourette’s syndrome (
42,
43) and of experimental CIN disruption producing repetitive behavioral pathology in mice (
45). CINs have been shown to differentially regulate D1R-expressing medium spiny neurons of the striatonigral (direct) pathway and D2R-expressing medium spiny neurons of the striatopallidal (indirect) pathway (
65,
66) via muscarinic acetylcholine receptors (
54,
67). The CINs interact with other neurotransmitter systems; they co-release glutamate (
68) and drive dopamine and GABA release from dopamine terminals (
69–
71). CINs can also indirectly inhibit medium spiny neurons via their up-regulation of GABAergic interneuron activity (
72–
75). Thus, inhibition of CINs may lead to dysregulated striatal output through several mechanisms.
Dysfunction of basal ganglia occurs commonly in other autoimmune encephalitides. Functional brain imaging studies show basal ganglia hypermetabolism in patients with autoimmune encephalitis (
76,
77). In anti–
N-methyl-
d-aspartate (NMDA) receptor encephalitis, loss of cortico-striatal inhibition due to antibody-mediated inhibition of GABAergic interneurons has been postulated (
78–
80). In another rare autoimmune disorder, stiff-person syndrome, anti-GAD65 (
81), and anti-amphiphysin (
82) autoantibodies disrupt GABAergic inhibitory synaptic activity, which may underlie the pathophysiology of this disorder. Our findings suggest that a similar mechanism may be at play in PANDAS, with antibody binding to interneurons leading to an imbalance between excitatory and inhibitory synaptic transmission in the basal ganglia and thus to clinical symptoms. Importantly, we have not shown antibody binding to CINs to be specific to PANDAS; indeed, since PANDAS is a clinical entity and its diagnosis is not based on a clearly defined underlying pathophysiology, we think it unlikely that elevated CIN binding will correspond precisely with clinical diagnosis. This binding pattern may not be seen in all patients with a PANDAS diagnosis (as suggested by the overlap of PANDAS and control values in
Figure 1), and a similar pathophysiology may contribute to other neuroinflammatory conditions. Systematic examination of interneuron binding across psychiatric and neurological diagnoses is an important future direction; such an examination will be facilitated by the identification of the associated molecular targets.
In this study, we tested PANDAS IgG binding to fixed mouse striatal slices and to human brain tissue, ex vivo. This differs from the in vivo infusion used in our pilot work (
47). These two strategies ask subtly different questions. In vivo antibody infusion demonstrates that antibodies in PANDAS sera can bind to CINs in intact brain, under physiological conditions, over days. The ex vivo approach used here is less chronic, examining binding after an overnight incubation (
Figures 1,
2, and
4) and functional effects after only 1 hour (
Figures 3 and
5; see also Figures S12 and S13 in the
online supplement). It allows better control of IgG concentration, which may vary between animals in vivo. It is less laborious, which allows for analysis of larger numbers of samples and the inclusion of additional controls. Importantly, the ex vivo approach permits, for the first time, examination of the functional effects of PANDAS serum on CIN activity (
Figures 3 and
5; see also Figures S12 and S13).
We also used an automated method to quantify IgG binding to CINs and other neuronal types (see Figure S2 in the
online supplement), which contrasts with the blinded manual counting used in our pilot work (
47). This allowed us to count an order of magnitude more cells than previously. On the other hand, the effect size of the elevated binding of IgG to CINs in our pilot work (d=8.1) was much higher than that we observed here (d=1.8 in the first cohort and somewhat less in the others). The use of standard fluorescent imaging rather than confocal imaging, which allows us to count more cells at lower magnification, leads to higher background, which doubtless contributes to this smaller (though still large) effect size. Lower background in the in vivo work may also derive from the more chronic serum administration and the 5-day clearance period after antibody infusion (
47), which may produce a better signal-to-noise ratio. Additionally, in our previous work, we categorized individual cells as either positive or negative for IgG, which is an inherently nonlinear approach and may amplify between-group effects. The finding of qualitatively identical results using these two experimental approaches, despite these differences, increases confidence in the finding.
There are, of course, several limitations to this study. The sera are as clinically homogeneous as we could achieve, especially in the first two patient cohorts, with rigorous PANDAS diagnosis and a narrow age range. These design choices were made to maximize our ability to find a pathophysiological signal in a small, homogeneous sample, which can subsequently be investigated in more heterogeneous populations. This approach has been fruitful in investigations of other autoimmune neural pathologies (
83). We examined binding to both mouse and human striatal tissue, but in both cases this tissue was from adults; it is possible that more or different binding would be seen on pediatric tissue. We focused on a specific brain region (the striatum) and on specific cell types, based on a priori considerations; it is possible that a more comprehensive survey would additionally reveal elevated binding by PANDAS IgG to other cells in other brain regions. And we focused on the ability of IgG to bind to interneurons but have not addressed the question of how these antibodies could access the brain in patients; this is an important, though distinct, topic for future research.
While the finding of elevated IgG binding to CINs documented here represents an exciting new insight into possible pathophysiological mechanisms in PANDAS, it would be premature to consider this a clinical assay. Overlap between PANDAS and control groups is substantial (
Figure 1B), and the specificity and prognostic utility of elevated CIN binding remain to be established. Development of a diagnostic assay based on these results may require the identification of the molecular target(s) of antibody binding to CINs. This is an important future direction. Ultimately, the identification and replication of specific binding targets, and clarification of their relationship to particular patterns of symptoms or of treatment response, may help clarify the diagnostic landscape in PANDAS and PANS, in addition to providing clinically useful laboratory tests.
In conclusion, we found that PANDAS antibodies showed elevated binding to striatal CINs but not several other neuronal types (
47), declined in parallel with symptom improvement after IVIG treatment, and reduced the activity of CINs—and perhaps altered their response to several neurotransmitters—ex vivo. This supports a novel hypothesis for the pathophysiology of PANDAS, which in turn suggests important directions for future research. Identifying cellular targets of pathogenic antibodies in PANDAS holds promise for validation of the diagnosis and for the identification of molecular targets for future treatment development.