Obsessive-compulsive disorder (OCD) is a common, chronic, and oftentimes disabling disorder characterized by unwanted and distressing thoughts (obsessions) and repetitive behaviors that the individual feels driven to perform (compulsions) (
1,
2). Compulsions can be either overt acts or mental rituals that typically serve to reduce distress engendered by the obsessions. A cardinal feature of OCD is that patients retain insight (to variable degrees) into the irrationality and excessiveness of their obsessive-compulsive (OC) behaviors. OCD affects 2%−3% of the U.S. population (
3) and is responsible for substantial functional impairment (
4,
5) and increased risk of early mortality (
6). The only established first-line treatments for OCD are cognitive-behavioral therapy with exposure/response prevention (ERP) (
7,
8) and serotonin reuptake inhibitor medications (SRIs) (
8–
12). Approximately 25%−40% of patients fail to respond to either modality (
13,
14), and few patients experience complete symptom resolution (
15). Beyond SRI monotherapy, the only medication approach for OCD with substantial empirical support is antipsychotic augmentation, particularly in patients with a comorbid chronic tic disorder (
16–
18).
Our incomplete understanding of the neurobiology of OCD has hampered efforts to develop new treatments or enhance extant interventions. As there are numerous clues to the biological underpinnings of OCD symptoms, armed with new technologies and tools in the lab and clinic, we may be on the threshold of testing hypotheses that will lead to optimization of existing treatments and discovery of new medications and devices that will alter the outcomes for patients with refractory OCD. In this review, we will not attempt to survey the whole field, rather we concentrate on several promising areas of research that may help elucidate the pathophysiology of OCD and advance treatment. We begin with what is currently known about the heritability and genetics of OCD.
OCD as Familial And Heritable
Twin and family aggregation studies support a significant genetic contribution to OCD and related disorders (
19). For example, based on a study of 5409 twin pairs (
20), higher concordance rates in monozygotic versus dizygotic twins resulted in an OCD heritability estimate of 48% (
19). Population-based studies have confirmed substantial heritability in OCD (
21). A multigenerational family clustering study of nearly 25,000 individuals with OCD—identified through the Swedish national registers—found that the risk for OCD among relatives increased according to the degree of genetic relatedness to the proband (
21). Shared environmental factors did not contribute additional risk for OCD (
21). Familial risk of OCD was previously reported as higher for probands with childhood-onset OCD compared with adult-onset probands (
22); however, the aforementioned population-based study did not find significant differences between these groups (
21). Based on their family data, Mataix-Cols et al. estimated the genetic contribution to OCD as approximately 50% (
21). A recent large study of individuals from the Swedish National Patient Register showed that 44%−48% of the total variance in risk for OCD is due to direct genetic effects, suggesting that the inflation of heritability from twin studies was modest (
23). However, after accounting for maternal effects, the estimate of heritability was 35% (95% CI: 32.3%−36.9%).
Maternal effects are influences on the offspring phenotype that result from maternal phenotypes and as such can be partitioned into genetic maternal effects and environmental maternal effects. Mahjani et al. (
23) showed that genetic maternal effects accounted for 7.6% of the total variance in risk for OCD, while shared environmental maternal effects had little or no effect on risk. Assortative mating among individuals with OCD has been reported in the literature (
24,
25), meaning that individuals with OCD choose a partner with OCD more frequently than expected under a random mating pattern. Assortative mating is estimated to inflate direct genetic effects by 4%−5% (
23).
Genetic Studies
Numerous candidate gene association studies in OCD have failed to deliver reproducible results (
19). The majority of these studies have examined genes associated with serotonin, dopamine, and glutamate containing pathways, representing the neurotransmitters most often implicated in the pathophysiology and treatment of OCD (
26,
27). Several candidate genes have also been identified through studies of animal models of OCD-like behaviors (e.g., excessive self-grooming alleviated by fluoxetine), including SAPAP3 and SLITRK5 (
28). The strongest OCD candidate gene to date is SLC1A1, which encodes the neuronal glutamate transporter EAAT3 (
29). Several gene variants influencing SLC1A1 expression have been associated with OCD (
19). Thus far, SLC1A1 has not acquired the stringent level of statistical evidence to be considered a definitive risk gene for OCD.
Two genome-wide association studies (GWAS) have been conducted to date, each involving about 1,400 cases (
30,
31). Neither study identified single-nucleotide polymorphisms (SNPs) associated with OCD at genome-wide significance level, nor did a meta-analyses of the two studies (
32). However, the Psychiatric Genomics Consortium (PGC) is currently working on a substantially larger meta-analysis that will include at least 14,000 individuals with OCD and over 560,000 controls.
Complex and heterogeneous psychiatric disorders like OCD may involve multiple common variants of small effects (
33); accordingly, existing GWAS in OCD may lack the statistical power to detect loci that reach genome-wide significance. Another strategy is to search for rare structural variants with large effects. Several copy number variation (CNV) studies have been conducted in patients with OCD (
19). The IOCDF Genetics Collaborative conducted a cross-disorder study of CNV in OCD (N=1,613), Tourette’s syndrome (N=1,086), and control subjects (N=1,789) (
34). Although no global CNV burden was detected in OCD (or Tourette’s), they found a 3.3-fold increased burden of large deletions on chromosome 16p13.1, a region previously associated with other neurodevelopmental disorders (
34).
A substantial portion of the genetic contribution to OCD is still unknown (
35). Larger sample sizes are needed to identify both common and rare variants involved in risk for OCD. A large-scale, population-based, prospective study of the genetics of OCD and chronic tic disorders is currently in progress that addresses several limitations of prior studies (
36).
Accidents of Nature and Structural Brain Abnormalities
Although widely believed to result from the interplay of genetic and environmental factors, some cases of OCD may be traced to specific neurological etiologies (
19). Evidence for a role of the basal ganglia in the pathophysiology of OCD comes from case reports of “accidents of nature” such as Sydenham’s chorea (
37), von Economo’s encephalitis (
38), and ischemic events (
39,
40) in which insults to the basal ganglia, particularly the globus pallidus and caudate, produced OC behaviors.
The OCD Working Group within the Enhancing Neuro Imaging Genetics through Meta Analysis (ENIGMA) Consortium has been leading the effort to identify structural brain abnormalities in OCD. Analysis of 16 pediatric and 30 adult datasets found asymmetry of subcortical structures in pediatric, but not adult, cases with OCD (
41). In another study from ENIGMA, no detectable brain structural differences were identified in a comparison of MRIs obtained from 2,304 OCD patients and 2,068 healthy controls (
42). The two groups (patients versus controls) could not be separated on the basis of their brain structural scans until medication status was factored into the analysis (
42).
Few postmortem studies of patients with OCD have been published. However, two recent studies found evidence for abnormalities in the orbitofrontal cortex of patients with OCD compared with controls. One study found layer-specific reduced neuronal density in the orbitofrontal cortex of seven subjects with OCD (
43). This study is limited by small sample size and that diagnoses were made postmortem. Another study investigated expression of synaptic genes from several brain regions (e.g., orbitofrontal cortex and striatum) implicated in OCD (
44). The authors found evidence for lower excitatory synaptic gene expression in the orbitofrontal cortex of the subjects (N=8) compared with unaffected controls (
44).
Serotonin Hypothesis and Efficacy Of SRIs
Like medication treatment of other psychiatric disorders (e.g., chlorpromazine for schizophrenia [
50] and imipramine for depression [
51]), the initial finding that SRIs are effective in OCD was a result of serendipity coupled with keen clinical observation. The observation in 1975 that clomipramine was beneficial in OCD (
52) sparked interest in the role of serotonin in this disorder. In contrast to other tricyclic antidepressants (TCAs), clomipramine is a potent inhibitor of the serotonin transporter. Clomipramine was shown to be more effective in OCD than the TCA desipramine, which is predominantly a norepinephrine reuptake inhibitor (
53). Later, selective SRIs (SSRIs) such as fluvoxamine and sertraline were also shown to be superior to desipramine (
9,
54). A large series of randomized clinical trials (RCTs) confirmed that clomipramine and SSRIs (e.g., fluoxetine, fluvoxamine, sertraline, etc.) were superior to placebo (
9,
10,
55–
57), establishing these medications as the mainstay for pharmacological management of OCD.
Since SRIs have a broad spectrum of action in psychiatric conditions, including depression and anxiety disorders, their efficacy in OCD is not a distinguishing feature. What stands out is that SRIs are effective in OCD, whereas other antidepressants that do not bind with high affinity to the serotonin transporter are generally ineffective in OCD (
9). These drug response data led researchers to hypothesize that dysfunction in brain serotonergic systems might underlie the pathophysiology of OCD (
26). A number of studies into the role of the serotonin (5-hydroxytryptamine [5-HT]) system in OCD were launched in the 1980s and 1990s. Many involved pharmacological challenge paradigms with probes of serotonin function (
58,
59); however, the findings were either equivocal or conflicting (
58–
60). One surprising finding was that acute tryptophan depletion, which temporarily reduces brain 5-HT levels, did not induce elevations in OC symptoms in a cohort of OCD patients who were SRI responders (
61). In contrast, depression ratings were increased during this tryptophan depletion challenge (
61). Positron emission tomography studies with radioligands that bind to the 5-HT transporter or the 5-HT2a receptor have thus far not revealed consistent group differences between OCD subjects and matched healthy subjects (
62). To date, direct evidence for serotonergic abnormalities in OCD remains elusive.
Multiple drug trials of serotonin modulating agents (e.g., 5-HT1a partial agonist buspirone [
63], 5-HT precursor tryptophan [
64], and 5-HT3 receptor antagonist ondansetron [
65]) were conducted, mostly as adjuncts to SSRIs in partial or nonresponders, some with encouraging initial results. Unfortunately, subsequent RCTs failed to confirm efficacy of augmentation with serotonergic agents (
63,
64,
66,
67). Further testing of high-dose ondansetron may be warranted (
67). The preferential efficacy of SRIs in OCD remains a tantalizing clue about a role for serotonin in treatment OCD, but so far it has not yielded insights into pathophysiology nor led to new treatment approaches (
26). There is a lesson here about the pitfalls of inferring pathophysiology from treatment response data and the presumed mechanism of action of the intervention.
Conceivably, potent SRIs may act through an intact serotonergic system to compensate for a disturbance in a functionally coupled system that is more directly tied to the underlying neurobiology of OCD (
26). Several research groups have hypothesized that the therapeutic action of SRIs in OCD may involve dampening of activity in hyperactive orbitofrontal-subcortical circuits that drive OC behavior, possibly restoring balance between direct and indirect frontal-cortical pathways (see subsequent discussion of neurocircuitry) (
68). Serotonergic neurons originating in the midbrain include projections to the orbitofrontal cortex and ventral striatum (
69), two brain regions implicated in the pathophysiology of OCD (
68). Preclinical studies have shown that 8 weeks of chronic SSRI administration, but not other antidepressants tested, leads to desensitization of 5-HT autoreceptors on axons terminating in the orbitofrontal cortex (
70).
Learning Theory and Efficacy of CBT
Learning theory models of OCD gained influence as a result of the success of cognitive behavioral therapy (CBT) (
71) and the growth of cognitive neuroscience (
72). Exposure and response prevention therapy is the first-line psychotherapy for OCD, having demonstrated large treatment effects (
14,
73). This approach originated on a conceptual framework emphasizing the salience of intrusive thoughts that motivate distress reducing, albeit problematic, behavioral responses (
74). Although nearly all individuals experience thoughts that share content with obsessions (
75), obsessions and associated distress result from cognitive misattributions of intrusive thoughts (e.g., equating thoughts with intent or action) that lead to a benign thought being interpreted as highly salient and dangerous. Such obsessional distress motivates compulsions or avoidance to reduce distress and prevent the undesirable outcome from occurring. Compulsions/avoidance are negatively reinforcing and prevent the person from realizing that the feared outcome was unlikely to occur and, if it had, the person could cope effectively. ERP directly targets this cycle and involves gradual and systematic exposure to distress-evoking stimuli while refraining from distress-reducing rituals or avoidance. However, the extent to which ERP informs the underlying pathophysiology of OCD remains unclear.
Mechanistically, the cognitive-behavioral model of OCD to understand symptom development, maintenance, and treatment is based on fear acquisition and extinction (
76). Conditioned fear occurs when an affectively neutral stimulus (conditioned stimulus [CS]) is associated with an aversive unconditioned stimulus (US), e.g., a person with unwanted intrusive beliefs that an otherwise affectively benign steak knife (originally the US) could be used to harm a loved one on impulse, thereby eliciting distress. After development of the conditioned relationship, the CS (i.e., knife) elicits a conditioned response (CR) of fear/distress. Many OCD patients further generalize these learned associations to other stimuli (e.g., sharp objects) (
77). Extinction involves the process of new learning in which repeated exposure to the CS in the absence of the feared outcome (e.g., illness/death) and/or engagement in safety behaviors (e.g., avoidance, compulsive rituals) results in an attenuated CR. Importantly, this process forms new learning of a CS/no US association that inhibits expression of the existing CS/US association and becomes more robust with repeated pairings (
78). A growing literature suggests that individuals with OCD differ from controls in the manner in which CS/US associations are developed, maintained, and extinguished (
79–
83), which may have implications for understanding treatment response.
Studies examining neural correlates of ERP response are another method of understanding the neurobiology of OCD. Yet, the literature has yielded inconsistent findings likely due to differing imaging protocols, ERP protocols, small sample sizes, and patient characteristics (e.g., medication status, treatment history). Overall, the cingulo-opercular and orbito-striato-thalamic networks have been implicated, supporting contemporary circuits models of OCD (
72). The cingulo-opercular network, which includes the dorsal anterior cingulate cortex (ACC) and anterior insula/frontal operculum, mediates attentional control (
84). A meta-analysis of fMRI studies found cingulo-opercular hyperactivity during error processing tasks in OCD (
84). With respect to orbito-striato-thalamic networks, multiple functioning imaging studies have found orbito-striatal hyperconnectivity in OCD at baseline that normalizes during treatment (
85). For example, post-ERP reductions in OCD symptom severity have been associated with decreased metabolism of the caudate nucleus (
86,
87) and thalamus (
88); however, effects of ERP on ACC have been mixed (
88,
89). Functional magnetic resonance imaging (fMRI) studies comparing pre- and post-ERP found decreased resting-state functional connectivity between left dorsolateral prefrontal cortex (dlPFC) and superior temporal gyrus, cuneus, and right precuneus (core component of the default-mode network [
90]) that correlated with reduced OC symptoms (
91). Another fMRI study in children with OCD found decreased recruitment of the dlPFC and left parietal cortex during an executive functioning task that normalized after ERP and correlated with symptom improvement (
92). Recently in a sample of 12–45-year-old patients with OCD, greater pretreatment activation in two networks—orbito-striato-thalamic during reward processing and cingulo-opercular during cognitive control—was associated with better treatment response to ERP (
93). Norman et al. (
93) advances the field by demonstrating the specificity of activated neural regions to ERP, suggesting potential biomarkers that may facilitate more personalized intervention.
Attempts to translate findings to improve treatment outcomes have yielded variable results. One approach involves targeting the
N-methyl-
d-aspartate (NMDA) receptor in the amygdala, which is critically involved in fear extinction, with the NMDA partial agonist
d-cycloserine (DCS). While individual studies have mixed findings, mega-analytic results of studies of DCS augmentation of exposure therapy in OCD and anxiety disorders demonstrated a small effect relative to placebo augmentation (−0.25) (
94) that could be further enhanced with optimized dosing and timing parameters (
95). While the potential of DCS augmentation has not been fully actualized, it represents an attempt to link ERP and the neurobiology of OCD by considering basic science research on the molecular mechanisms of fear extinction.
Glutamate: From Neurobiology to Treatment
A role for the glutamatergic system in the neurobiology of OCD has been gaining traction as a result of emerging imaging data (
96), genomic studies (
97), biochemical studies of cerebrospinal fluid (CSF) (
98,
99), and animal models of aberrant grooming behavior (
100) (for a detailed review of the glutamatergic hypothesis of OCD, see Pittenger et al. [
27]). These findings have stimulated interest in examining the efficacy of medications that modulate glutamate function (
10). Most of the medications tested act to reduce glutamatergic activity. Involvement of glutamate in the pathophysiology of OCD is highly compatible with circuit-based theories of OCD that will be subsequently discussed.
The ubiquitous amino acid glutamate serves as the primary excitatory neurotransmitter in the adult brain (
27). Along with the inhibitory neurotransmitter gamma-aminobutyric acid (GABA), glutamatergic projections participate in the connections forming the cortico-striato-thalamo-cortical (CSTC) circuit that has been implicated in the pathophysiology of OCD. Regulation of glutamatergic activity is complex. Glutamate released from the presynaptic membrane diffuses and binds to ionotropic receptors (NMDA, α-amino-3-hydroxy-5–4-isoxazoleproprionic acid [AMPA], and kainite) and metabotropic glutamate receptors (mGluR3 and mGluR5) located postsynaptically and presynaptically and to transporters (excitatory amino acid transporter [EAAT] 1 and EAAT2) on glial cells, which remove glutamate from the extracellular space.
One of the first glutamate-modulating agents to be tested in the treatment of OCD is riluzole (
27), a medication with Food and Drug Administration (FDA) approval for amyotrophic lateral sclerosis (ALS). It exerts a number of actions on glutamatergic activity, including inhibiting presynaptic release of glutamate, direct effects on ionotropic receptors, and increased expression and function of EAAT2, which may increase clearance of glutamate (
101). Several positive open-label studies of riluzole augmentation were reported among adults and adolescents with treatment-resistant OCD (
27). These were followed by two RCTs of adjunctive riluzole versus placebo in adults (
102) and children (
103) who had inadequate responses to SRIs alone. Neither study found significant group differences on the primary endpoints, but a secondary analysis showed a trend favoring riluzole in adult outpatients (
102). Although generally well tolerated, riluzole is associated with elevated transaminase levels, and pancreatitis occurred in two children with OCD (
103). Troriluzole (formerly BHV-4157) is a prodrug conjugate of riluzole that bypasses first-pass metabolism, a property that confers improved bioavailability and, potentially, lower risk for hepatoxicity (
104). Troriluzole is currently undergoing evaluation as an adjunctive medication in adults with OCD (NCT03299166).
Memantine is a low-affinity noncompetitive NMDA blocker with an FDA indication for treatment of Alzheimer’s disease. A series of open-label studies showed benefit of adjunctive memantine in patients with OCD (
10). In a case control study of 44 patients treated at McLean Hospital, 22 who received memantine showed greater improvement (
105). A pair of RCTs from a different center claimed that memantine was superior to placebo either as an adjunct or monotherapy; response rates were higher than expected: drug 81% versus placebo 19% (
106,
107). The validity of these findings, and a subsequent meta-analysis of memantine in OCD (
108), has been questioned (
109). Of the four studies included in the meta-analysis, only one enrolled SRI-refractory patients (
109), dropout rates were high, and completer analyses (instead of more rigorous intent-to-treat analyses) were performed in each RCT (
109). Additional controlled trials of memantine by independent research groups are warranted.
Ketamine, a noncompetitive antagonist at NMDA receptors, is a rapid-acting antidepressant (
110). Esketamine, the s-enantiomer of ketamine, is FDA-approved for treatment-resistant depression as a nasal spray. An open-label study of intravenous ketamine was conducted in 10 patients with treatment-resistant OCD (
111). Both OC and depressive symptoms improved significantly at 3 days postinfusion compared with baseline, but none of the subjects met criteria as an OCD responder, defined as a 35% reduction on the Y-BOCS (
112). Another research group performed an RCT of intravenous ketamine versus placebo in 15 medication-free subjects with OCD (
113). At one-week postinfusion, 50% of the ketamine group were OCD responders compared with 0% of the placebo-administered group. These positive findings reinforce the need for larger confirmatory RCTs.
N-Acetylcysteine (NAC) is a prodrug of the amino acid cysteine that is available over the counter and generally well-tolerated (
114). NAC modulates glutamate via glial cystine/glutamate exchange (
115) and increases cellular production of the antioxidant glutathione. NAC is used as an antidote for acetaminophen poisoning. A placebo-controlled RCT of adjunctive NAC (3,000 mg daily) was carried out in 40 adults with treatment-resistant OCD (
116). No significant between-group differences were found at the conclusion of this 16-week trial (
116) nor in a small parallel study in youth (
117). A secondary analysis suggested that NAC may reduce anxiety (
116).
The nonessential amino acid glycine functions as an allosteric agonist at NMDA receptors (
118). Several compounds that influence glycine availability, including glycine itself, or act on glycine receptors or transporters, have been investigated in OCD. Efficacy of adding glycine to patients with OCD (N=24) stabilized on SRIs was evaluated in a double-blind, placebo-controlled trial (
119). The high dropout rate because of gastrointestinal side effects from glycine confounded interpretation of the findings (
119). Sarcosine is an endogenous inhibitor of the glycine transporter type 1 (GlyT-1) and is thought to increase levels of glycine (
120). In an open-label trial of adjunctive sarcosine, eight (32%) of 25 subjects were responders (
120). A multicenter RCT (N=99) of the specific GlyT-1 inhibitor bitopertin was performed in patients with SSRI-refractory OCD (
121). Bitopertin failed to separate from placebo in augmenting response to ongoing SSRIs at week 12 of the double-blind phase (
121). DCS is a partial agonist at the glycine/
d-serine site on NMDA receptors. Its potential to augment ERP by enhancement of extinction learning was discussed earlier.
Neurocircuitry of OCD
A confluence of evidence—from studies of brain imaging, cognitive-affective neuroscience, neuromodulation, and animal models—suggests that OCD is a prime example of a disorder borne of dysfunction not within a single region in the brain but rather within networks of brain regions (
122). One productive way of conceptualizing these networks and studying their dysfunction in OCD is based on the motif of CSTC loops and related regions (
123). Using this framework, functional brain imaging studies in OCD have generally demonstrated consistent results (
123). Both positron emission tomography (PET) (
124) and fMRI (
125) have shown increased activation in regions of the orbitofrontal cortex (OFC), anterior cingulate cortex (ACC), and portions of the basal ganglia (particularly caudate nucleus) in the symptomatic state compared with healthy controls (
85). These areas of abnormal activation tend to normalize with successful treatment upon repeated testing, whether with medications (↓ACC, ↓caudate, ↓OFC) or ERP (↓caudate) (
85,
87,
126). Other effective treatment modalities are also associated with reductions in brain activity compared with baseline in OCD: deep brain stimulation (DBS) (↓ACC, ↓PFC) (
127,
128), neurosurgical lesions (↓ACC, ↓caudate, ↓PFC, ↓thalamus) (
129,
130), and transcranial magnetic stimulation (TMS) (↓ACC, ↓OFC, ↓PFC) (
131,
132).
One of the most important technical and conceptual advances in circuit-based theories of OCD has been the change in focus from static regions of interest to interrogation of functional networks subserving different cognitive or behavioral functions germane to the symptomatology of OCD (
133–
140). The seminal work by Alexander et al. (
141) influenced OCD researchers to consider certain parallel segregated CSTC loops, subserving different motor or cognitive functions, as the neuroanatomical substrate for OC behavior (
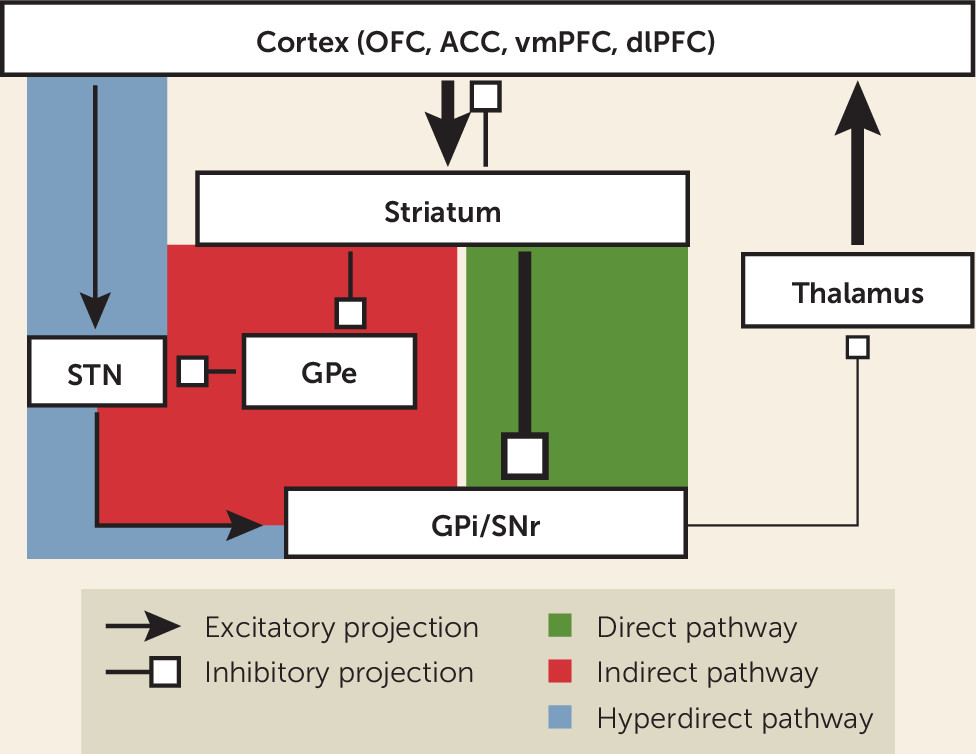
85). The canonical model of a CSTC loop consists of projections from a specific region of the frontal cortex that terminate in the striatum, and then travel by either a “direct” (net excitatory) or “indirect” (net inhibitory) pathway through the basal ganglia to the thalamus, and finally returning via recurrent projections to the same cortical region to close the loop (see
Figure 1). The striatum is the largest structure of the basal ganglia and serves as its main input region. It is composed of the caudate, putamen, and ventral striatum (VS), which contains the nucleus accumbens (NAc). The main output structure of the basal ganglia is the globus pallidus interna/substantia nigra pars reticulata (GPi/SNr) complex, which largely projects to the thalamus. Other important relay structures within the basal ganglia are the globus pallidus externa (GPe) and subthalamic nucleus (STN).
Most projections within the basal ganglia, with the main exception of the STN, are inhibitory. Thus, the overall balance of inhibition and excitation is determined by relative contributions of direct and indirect pathway components. The direct pathway projects from the striatum to the GPi/SNr. Striatal inhibition of the GPi/SNr disinhibits the thalamus, producing net excitatory tone. In contrast, the indirect pathway projects from the striatum to the GPe, and then to the STN, before returning to the GPi/SNr and rejoining the common pathway (
85). These additional relays add one node of inhibition, resulting in a net inhibitory tone within the circuit. Revisions of this model paint a more complex picture of the organization of CSTC loops (
123), showing more overlap and functional integration between loops than previously appreciated and the importance of other interconnected structures such as the amygdala and hippocampus (
142).
Several research groups have hypothesized that excessive tone in the direct pathway (relative to the indirect pathway) would explain some of the neuroimaging findings and phenomenological features of OCD (
85,
143). According to this model, such an imbalance in activity between these pathways would bias the individual to selection of, and failure to inhibit, abnormal and repetitive behavioral sequences (
85,
143,
144). Hyperactivity of orbitofrontal-subcortical pathways has been found in both human imaging studies in OCD and mouse models of OCD-like behaviors is (
144). An influential study by Ahmari et al. (
145) used optogenetics in mice to test the effects of CSTC hyperactivation on behavior. Repeated stimulation (over multiple days) of OFC-ventral striatum projections induced increased grooming behaviors, which persisted after stimulation cessation. Chronic administration of fluoxetine reversed the aberrant grooming (
145). These data provide strong support for a role of OFC hyperactivity in the genesis of abnormal repetitive behaviors. Together, converging lines of clinical and preclinical evidence suggest that OCD involves dysfunction of limbic CTSC loops that include the OFC, vmPFC, ACC, and ventral striatum (
146).
Transcranial Magnetic Stimulation
Recently, the FDA cleared a form of repetitive transcranial magnetic stimulation (TMS), referred to as deep TMS (dTMS), for the treatment of OCD based on the results of a pivotal trial (
147). In this double-blind RCT of adults (N=99) who had limited response to previous treatments, 38% responded to the device compared with 11% who received sham treatment (
147). This device uses an H-shaped coil that was designed to reach deeper structures, 3 cm to 5 cm penetration, compared with electromagnetic stimulation depth of about 2 cm with conventional figure-8 coils (
148). The brain regions targeted with dTMS were midline structures, medial PFC (mPFC) and ACC, two brain areas thought to be hyperactive in OCD (
147).
Other studies of rTMS in OCD have targeted different cortical brain regions, including the dlPFC and OFC, with mixed results (
147). Dunlop et al. (
131) used rTMS targeting the dorsomedial PFC (dmPFC) to treat 20 adults with treatment-resistant OCD. Resting state fMRI (rsfMRI) was performed pre- and posttreatment. Ten subjects met stringent response criteria. Responders had higher dmPFC-ventral striatal connectivity at baseline (
131). Furthermore, dmPFC-rTMS treatment was associated with reductions in hyperconnectivity that correlated with improvement on the Y-BOCS (
131). These findings with noninvasive neuromodulation provide additional evidence for cortico-striatal hyperactivity underlying the symptoms of OCD.