These broad statements about the impact of gene discovery in ASD may ring hollow to some. Approaches to diagnosis, the ability to predict clinical course and natural history, and our therapeutic armamentarium have not changed markedly over this time. There is no question that the pace of progress is too slow for our patients and their families. Still, the understandable frustration should not obscure how different the field is today than it was a decade ago. The most recent reliable gene lists are extensive and growing, and the protein products of these genes point to a relatively small number of areas of biology where parallel advances in neuroscience and functional genomics are yielding profound insights. There is now replicable evidence pointing to a specific cell type, brain region, and developmental epoch that constitute one nexus of vulnerability for some of the highest-confidence ASD risk genes. And there are multiple plausible paths forward toward rational therapeutics that, while extremely challenging, are nonetheless radical departures from what was possible prior to the era of successful gene discovery in ASD.
A Decade of Successful Gene Discovery Has Clarified the Genetic Architecture of ASD
Judging from the sheer number of identified loci, the majority of progress in the genetics of common medical and psychiatric conditions over the past decade has resulted from genome-wide association studies (GWASs) aimed at characterizing classic polygenic inheritance—that is, the combined contribution of many small-effect polymorphisms (alleles) to disease risk. ASD has been a striking exception: here, the yield of studying individually rare (found in less than 1% of the general population) mutations has vastly outpaced findings with regard to common alleles. Recent investigations have identified more than 100 genes that meet rigorous statistical thresholds for association with the ASD phenotype (
6,
7), based predominantly on the finding of rare, spontaneous (de novo) germline mutations mapping to the genic portion of the genome and predicted to be deleterious to the function of the encoded protein (
Figure 1A,B).
Many of the earliest investigations that focused specifically on the potential role of de novo mutations in common or idiopathic forms of ASD involved the study of copy number variants (CNVs). These are submicroscopic alterations in chromosomal structure, including losses (deletions) or gains (duplications or amplifications) of segments of DNA, typically involving tens of thousands to several million nucleotides. It was not until the completion of the human genome project that it was appreciated that these were part of the normal complement of human genetic diversity (
8,
9), appearing in about 1% to 2% of the typically developed population. Shortly thereafter, the seminal observation was made that “large” de novo CNVs were more prevalent among ASD probands from families with only a single affected individual (simplex families) compared with families with multiple affected individuals (multiplex families) or compared with typically developing control subjects (
10).
This work and that of several other groups that quickly followed (
11–
14) represented a critical advance: 1) they pointed to CNVs carrying large effects as a group—as the increased rate in ASD was apparent in quite modest cohort sizes; 2) they demonstrated that the finding in affected individuals was not a consequence of nonspecific chromosomal fragility, but rather an accumulation of specific risk regions in the ASD population; and 3) with increasing cohort sizes over the ensuing several years, multiple specific ASD CNVs were confirmed, based on rigorous statistical thresholds (
11,
15–
18). These investigations also demonstrated that a loss or gain of a single segment of an individual’s genome, from either one of the two inherited chromosomes in a pair, was sufficient to confer risk—that is, there was no evidence that an individual needed to lose copies from both chromosomes or carry multiple de novo CNVs, or that the loss of a segment of DNA was uncovering a damaging mutation on the remaining intact chromosome. Moreover, studies identified ASD risk CNVs across the full range of intellectual ability among affected individuals (
18), challenging the conventional wisdom that these types of mutations would primarily be found to be responsible for global developmental delay, with ASD emerging as an epiphenomenon. Finally, there was widespread replication of the finding that both increases and decreases in copy number at precisely the same locus could be associated with a neurodevelopmental phenotype, sometimes involving the same psychiatric diagnosis.
In fact, some of the most striking and surprising observations from these early CNV studies related to genotype-phenotype relationships. For example, a CNV identified at chromosome 16p11.2 was one of the first to be associated with typical ASD (and turned out to be the most common risk CNV in idiopathic ASD cases) (
11,
15,
16). As simplex cohorts grew in size, studies confirmed that duplications and deletions of this region were independently associated with ASD risk (
17,
18). However, both increases and decreases in copy number in precisely the same region were also found to increase risk for a wide variety of phenotypes, including intellectual disability (
19), epilepsy (
20), obesity (
21,
22), and schizophrenia (
23). In fact, over time, as de novo CNV studies were extended across psychiatric research populations, it became increasingly difficult to identify any one of a growing list of ASD-associated structural variants that did not also confer some risk for another disorder or phenotype, ranging from schizophrenia to bipolar disorder to attention deficit hyperactivity disorder (ADHD) to specific language impairment to a leftward shift in IQ among carriers. The upending of expectations regarding a differential impact of DNA gains versus losses and the dramatic absence of phenotypic specificity, despite very large effect sizes, presented immediate conceptual challenges for the field, including for strategies aimed at illuminating biological mechanisms underlying ASD—a topic that will be the focus of the sections below.
Another critical contribution of the early work on ASD CNVs was the related development of patient resources (
24) and statistical frameworks (
18) that set the stage for a decade of success in the systematic identification of risk genes. In fact, the subsequent epoch of rapid progress in gene discovery was dominated essentially by a single experimental design—namely, looking for the recurrence of de novo rare mutations across the genome in simplex ASD—while relying on increasingly high-resolution genomic assays, increasingly sophisticated statistical approaches to evaluating recurrent mutations (
25,
26), and increasingly large patient cohorts. In this regard, the development of “next-generation” sequencing was a key advance, reflected in 2012 by four essentially simultaneous publications confirming that, similar to CNVs, there was a statistically significant increase in the rate of de novo genic point mutations (single-nucleotide variants [SNVs]) in simplex ASD probands compared with unaffected sibling control subjects or theoretical expectations (
1–
4).
In some senses, these reports constituted an incremental advance grounded in the knowledge of the contribution of de novo CNVs to ASD. However, these initial findings, leveraging the ability to sequence the coding segment of the genome (the exome), were nonetheless seminal in that they presented a clear path forward for the systematic identification of large-effect ASD genes—and subsequently the discovery of large-effect genes conferring risk for multiple psychiatric phenotypes, including ADHD (
27), Tourette’s syndrome (
28), obsessive-compulsive disorder (OCD) (
29), and schizophrenia (
30,
31).
The earliest ASD exome sequencing studies showed that, similar to CNVs, the increased rate of de novo SNVs could be detected in samples of less than several hundred families, pointing to large biological effects, and they demonstrated that the increased rate was found only for particular types of SNVs—those predicted to be damaging to the encoded protein. The strongest evidence was found for the most deleterious variants, referred to as putative loss-of-function or likely-gene-disrupting (LGD) mutations—those resulting in the insertion of a premature stop codon, frameshift mutation, or alteration of a canonical splice site. Importantly, in general, these LGD mutations were heterozygous (present only in one of two copies of a given gene) and were predicted to result in haploinsufficiency—a reduction by about half of the total expression of a given gene. In addition, from the outset, there was no evidence that as a group, probands with ASD were more likely than sibling control subjects to carry de novo coding mutations simultaneously in multiple different genes (
1), indicating that the risk to the individual was typically the consequence of a single damaging heterozygous de novo coding mutation.
Together these initial ASD DNA sequencing studies discovered a small number of novel risk genes, including
SCN2A,
CHD8, and
GRIN2B, and were also able to show, through modeling, that several hundred to a thousand additional risk gene “targets” of this type of mutational mechanism would be identified as cohort sizes increased. And as the number of ASD risk genes has accumulated, largely at the predicted rate (
6,
7), additional characteristics of the genomic architecture have been clarified. For example, LGD mutations have been found to be most likely bona fide ASD risks when they are identified in brain-expressed genes that are under strong evolutionary constraint. By evaluating how prevalent inherited damaging mutations are in a gene in the general population, one can discern how well it “tolerates” deleterious mutations (
32). If a gene has significantly fewer putative loss-of-function variants than would be expected by chance, it is likely that this is a consequence of the most damaging variations being removed by natural selection over generations. As a group, replicable de novo LGD risk mutations have been found to map to genes that are, in fact, markedly depleted for the expected number of likely gene-disrupting variants—a phenomenon that has also been found with regard to other neurodevelopmental phenotypes, including epilepsy, intellectual disability, and schizophrenia (
30).
In addition, as with CNVs, individual SNVs in the same gene have been found to confer risk across distinct psychiatric and developmental phenotypes: for example, for developmental delay, epilepsy, schizophrenia, ADHD, congenital heart disease, and OCD (
27,
29,
30,
33–
35). However, the phenomenon of a single risk gene being associated with disparate disorders does not appear to be as ubiquitous as has been described for CNVs. For example, a recent study of rare and de novo LGD SNVs in schizophrenia confirmed 10 high-confidence risk genes based on the finding of rare heterozygous deleterious mutations in highly evolutionarily constrained genes (
30). However, none of these were present in the recent lists of more than 100 ASD-associated genes identified using similar approaches (
6,
7). When the investigators expanded their search to the next 22 probable schizophrenia risk genes that did not meet their most stringent statistical threshold but still showed a false discovery rate of less than 5%, only three had been previously identified as high-confidence ASD genes (
6,
7). The list of schizophrenia rare risk alleles remains relatively small, and there is likely to be additional overlap as rare-variant-based gene discovery proceeds for both diagnostic categories. However, at present, these findings suggest that individual genes may carry differential risks for different psychiatric diagnoses. Supporting this notion, a recent large-scale analysis of rare and de novo mutations in ASD (
7) showed differential risk for specific genes between ASD and more global developmental delay. In short, while many genes identified so far carry risk for more than one disorder, there is increasing evidence for some degree of relative specificity, for example, for ASD, intellectual disability, schizophrenia, or epilepsy—a tantalizing observation for investigators hoping to develop therapeutic targets by linking specific molecular or cellular mechanisms to distinct disorders (
1,
34,
36,
37).
Before moving to a consideration of the impact of successful rare variant gene discovery for translational science, it is important to note that ASD GWAS efforts have recently been successful as well. A well-powered, rigorous analysis (
38) reported five alleles associated with ASD. As expected, this required a very large cohort (more than 18,000 cases) and confirmed that the individual effects of risk alleles were quite small. Nonetheless, this avenue of investigation holds considerable promise for advancing the understanding of ASD. For example, it is very likely that the variability of outcomes for single large-effect de novo mutations is at least in part attributable to the interaction of rare and common alleles. Dissecting this interaction—with regard to both risk and resilience—is an increasingly tractable experiment and one that could have a profound impact on our understanding of natural history, treatment response, and the development of novel therapeutics.
Viewing just the current scorecard of gene discovery in ASD based on rare versus common alleles, it is tempting to conclude that rare SNVs and CNVs underlie the majority of ASD cases. They do not (
39,
40): for all patients presenting for evaluation of idiopathic ASD, positive yields for current genetic testing fall around 5%−10% in unselected populations, although this yield is anticipated to increase as the number of identified ASD risk genes and regions grow. However, the denominator in this equation is also critically important: there are multiple phenotypic features that significantly increase the likelihood of identifying a rare contributing mutation in a patient presenting to clinic, including the presence of seizures, a larger number of unaffected siblings (increasing the likelihood that the pedigree is truly simplex), and female sex (
6). In addition, lower IQ increases the likelihood of finding a relevant CNV or SNV, although, as noted, these mutations are not restricted to patients with low IQ or meeting diagnostic criteria for intellectual disability.
Despite the remarkable track record, exome analyses have already begun to give way to studies relying on whole-genome sequencing. This is an expected transition given that whole-genome sequencing captures essentially the full range and extent of genomic variation in a single assay—and costs have now fallen sufficiently to make this approach viable for large-scale studies. Early efforts have reported suggestive findings with regard to the contribution of rare noncoding mutations to ASD, although at present, research cohorts are too small to confirm the association of individual rare mutations in the noncoding portion of the genome, while still conforming to the statistically rigorous standards that have empowered a generation of successful human genetic studies (
41,
42).
Finally, it is important to emphasize the “looking under the lamppost” character of this summary of recent insights into the genetic architecture of ASD. The foregoing describes a remarkable trajectory for successful gene discovery based on the particular ease of detecting rare de novo mutations that damage gene function and the fortuitous discovery that haploinsufficiency plays a role in ASD risk. However, these methods, while in some sense hypothesis free, do confer inherent ascertainment biases. For example, studies that have included the search for
inherited rare variations have found that these can also confer risk, again particularly when mutations are found in evolutionarily constrained genes. Moreover, while it is relatively straightforward to predict mutations that result in a decrease or loss of function, it remains far more difficult to detect gain-of-function SNVs without a deep understanding of a given gene. Based on the finding that both losses and gains in chromosomal structure carry risk for neurodevelopmental phenotypes, it is quite plausible that gene discovery yields will accelerate as our ability to interpret—writ large—a wider range of coding variation deepens. In addition, there is definitive evidence that there are recessive forms of ASD, that somatic mutations and a variety of noncoding variations contribute to risk, and, as noted, that common variants carry the lion’s share of population risk for ASD (for recent reviews, see
5,
43). Consequently, it is an important caveat that despite the ample harvest of definitive molecular clues, there is far more yet to learn about the genetic contributions to ASD and that the nature of the methods for gene discovery may, to some degree, dictate what aspects of ASD biology are illuminated.
Leveraging Large-Effect Genes to Unravel the Biology of ASD
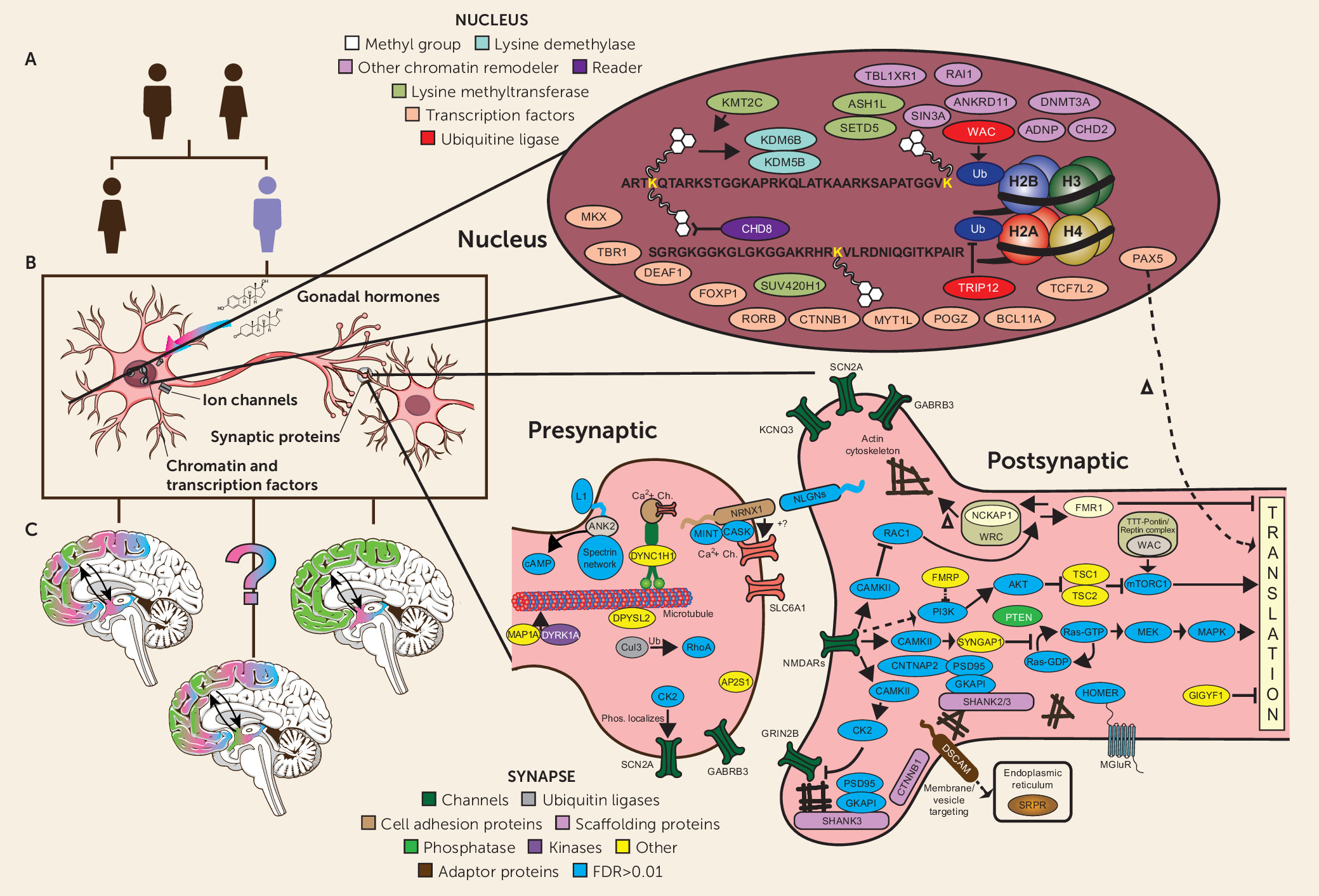
It is striking that despite the very high degree of locus heterogeneity—that is, the number of different genes that can lead to the ASD phenotype—the earliest studies of de novo mutations all observed that a significant number of risk genes and regions pointed prima facie to one of two major areas of biology: synaptic structure/function and the modification of chromatin (
3,
18,
44–
49) (see
Figure 1B). With increasingly large cohorts, one study after another has reinforced these findings and identified additional areas of putative functional intersection, or convergence, among ASD risk genes. These include (but are not limited to) relative enrichment of genes expressed in fetal brain, of transcription factors and RNA binding proteins, and specifically of targets of the Fragile X Mental Retardation Protein (FMRP) (
43,
45,
46,
50)—a finding that is a striking reminder that there is still much to learn at the intersection of common forms of ASD with so-called syndromic intellectual disability syndromes.
At one level, the ability to place the proteins encoded by ASD risk genes into relevant functional contexts is quite satisfying. At the same time, increasingly detailed diagrams such as the one shown in
Figure 1B obscure some fundamental challenges in seeking to clarify ASD pathology as a step toward the development of more effective treatments. While it is now relatively straightforward to characterize aspects of the biology of ASD risk genes, either pointing to annotated descriptions of “what they do and what they bind to” or via conducting experiments to determine what happens when ASD mutations are introduced into a range of model systems (
43), it is far more difficult to determine what aspects of observed biology are specifically relevant to the development of social disability. Indeed, the distinction between predicting or observing ASD-gene-related biology and identifying their pathological mechanisms in human disorders has become a major preoccupation for those searching for therapeutic targets (
43).
At issue is the multidimensional pleiotropy of genes, especially genes involved in the development of the human brain. For example, genes often encode multiple isoforms of a given protein, each of which may not do only one thing: annotation schemes may focus on the most well-known function of the most common isoform, but mechanistic pleiotropy is the rule rather than the exception. Moreover, the biological function of a given gene may be highly dependent on when and where a given protein isoform is being expressed. For example, a single gene may subserve cell proliferation at one point in development and synaptic transmission at another. Of course, this complexity is particularly pronounced with regard to the development of the human brain. This is an organ with extraordinary cellular diversity that exhibits tremendous biological dynamism, especially in prenatal and early postnatal development (
51,
52).
In short, a single gene in a list of ASD risk genes is likely to lead to the production of more than one protein isoform, each one of which may be found in more than one cell type, which in turn may contribute to more than one micro or macro circuit underlying a potentially wide range of perceptions and behaviors, which may vary depending on which model system is being examined—and this complexity has additional dimensions, as noted above, including developmental time course, and, in the case of ASD, sexual dimorphism. In short, each of the many ASD risk genes potentially presents a potentially impenetrable one-to-many-to-many problem.
Multiple laboratories have taken on this question by focusing on the notion of convergence across dimensions (
43,
50,
53). This is a logical extension of asking whether there is overlap among the described functions of protein products of diverse risk genes, built on the hypothesis that somewhere along the path from a diverse set of risk genes to a clearly definable clinical phenotype, there are likely to be shared causal pathways. As the amount of data regarding the molecular and cellular characteristics of brain development has grown dramatically, so too has the ability to investigate whether disparate high-confidence ASD risk genes demonstrate spatiotemporal convergence, not just functional intersection—in short, whether there is convergence in
when and
where there may be a point of vulnerability in the developing brain. Many of these analyses combine gene lists with “omics” data sets that capture the dynamic trajectory of brain development, including in the human.
For example, in some of the earliest forays into this approach, two studies took the highest-confidence ASD genes at the time and combined these with emerging data on the developmental trajectory of gene expression in human brain (
54,
55). Despite using divergent methods, different expression resources, and some different model systems, these two studies, appearing in the same issue of the journal
Cell, found that ASD risk genes showed greater convergence than expected by chance in mid-fetal cortical development and in excitatory neurons. While they diverged with regard to whether the point of greatest intersection was upper or deep layer neurons (or both), what was more striking was the agreement in these, and subsequent studies, in showing vulnerability in pyramidal cells in mid-fetal prefrontal cortical development (reviewed in
43,
56,
57) for a specific subset of risk genes. Not surprisingly, as both the number of high-confidence ASD genes has grown and the amount and resolution of data on the molecular landscape of the developing brain has expanded, additional points of potential convergence have begun to emerge. At the same time, multiple laboratories have turned to investigating convergence among ASD risk genes, leveraging a wide range of model systems, including induced pluripotent stem cell and brain organoids. Together these approaches offer a promising avenue to begin to differentiate the rapidly growing knowledge base regarding the multiple consequences of a given mutation in an ASD gene from those aspects that may more specifically underlie pathobiology.
Despite the many insights provided by recent ASD gene discovery, even a preliminary answer for one of the most striking and obvious aspects of ASD pathology remains obscure: How do the molecular and cellular pathways implicated in ASD pathobiology, which themselves are common to both sexes, intersect with those mediating the sexual differentiation of the mammalian brain to produce the male bias in incidence in these disorders (
7,
58,
59)? Studies of both CNVs and SNVs show a greater burden of damaging mutations in females compared with males. Similarly, studies have generally shown a higher incidence of ASD in families of affected females. Both observations point to the possibility of inherent resilience to genetic risks among females, often referred to as the female protective effect (
59,
60–
62). For example, if girls are more resilient to polygenic ASD risk, the presence of an affected female in a pedigree would be expected to identify those families carrying the greatest overall amount of risk, which would then tend to be manifested in greater numbers of relatives meeting diagnostic thresholds.
In mammals, differences in gene expression arising from multiple mechanisms, including as a consequence of mapping to the sex chromosomes or through the actions of gonadal hormones, mediate the sexual differentiation of the brain (
59). Accordingly, the earliest efforts to identify genes conferring risk for idiopathic ASD were accompanied by some speculation that, similar to intellectual disability, there might be an accumulation of risk loci present on the sex chromosomes. In support of this notion, studies of sex chromosome aneuploidy in humans and in rodents reveal effects of sex chromosome dosage on varied cognitive processes, including social, language, motor, and spatial skills, as well as brain volume in specific cortical and subcortical regions (
63–
65). With regard to the individual sex chromosomes, a small number of Y-linked genes are expressed in the brain, but their function in the nervous system remains unknown (
66,
67). In contrast, the X chromosome appears to be enriched for genes that are expressed in the brain, and X-linked mutations are often associated with intellectual disability—syndromes that are more prevalent in boys than girls (
68,
69). Moreover, the earliest success in rare-variant ASD genetics (
48) focused on the contribution of X-linked chromosomal abnormalities in girls and discovered rare LGD mutations in the genes
NLGN4 and
NLGN3, both mapping to the X chromosome.
However, with the characterization now of more than 100 ASD genes, there is not substantial evidence for a marked accumulation of ASD risk on the X chromosome. In short, mapping the genomic coordinates of risk genes so far has not made a substantial contribution to our understanding of sexual dimorphism in ASD—although methods for studying the sex chromosomes have lagged behind autosomes, so there could be a more substantial contribution that has so far eluded detection.
An attractive alternative explanation has focused on differential gene expression between the developing male and female brains. As noted, analyses of gene expression related to ASD-associated genes have implicated mid-fetal cortical development (
54,
55). It is important to note that these early analyses relied on available expression data that was enriched for human postmortem cortical tissue, suggesting that the sensitivity to detect other points of convergence was limited. Moreover, even in these studies, subcortical regions also showed some evidence for convergent risk (
56). Importantly, the developmental epoch most strongly supported to date in studies of ASD pathology corresponds to the developmental periods of testosterone surges observed in mid-gestational humans and perinatal rodents (
70), suggesting a critical window during which gonadal hormones act to mediate sexual differentiation of the brain. Together these observations suggest that this may reflect an important insight into the “when,” of sexual dimorphic risk, but still leaves open the question of “where”—specifically, what regions of the brain are affected by steroid hormone receptor (SR) transcription factors to control sex-typical patterns of development, neural activity and behavior, and presumably sex differences in pathology. Of note, studies in humans reveal only subtle sex differences in gene expression in cortical tissue (
51,
52,
66,
71), and so far there is no evidence that subsets of ASD risk genes show differential risk between males and females, which might be expected if the expression of these genes were substantially influenced by sex. Thus, the question remains as to where the mechanisms that mediate sexual differentiation of the brain intersect with those that mediate ASD pathogenesis.
One possibility is that sex differences in the brain and ASD pathogenesis manifest in different brain regions and intersect on the basis of long-range connectivity. For example, many subcortical regions identified by the cellular expression of SRs (reviewed in
59) show significant connectivity both between each other and with cortical regions that control social behaviors in particular (
59,
72,
73). Disruptions to cortical function resulting from ASD pathogenesis may then act upstream or downstream of sex-specific patterns of connectivity or activity, for example, in the hypothalamus, which is enriched in SRs, displays significant differences in gene expression, cell number, and connectivity between males and females, and controls sex-typical displays of behavior (
74,
75) (
Figure 1C). It is also possible that ASD risk and sexual dimorphism intersect at the cellular level in specific populations in the cortex that have yet to be identified, as subtle spatially and temporally dynamic differences in gene expression can be difficult to detect and may yet be found to confer important effects with regard to either male risk or female resilience. Similarly, the alternative remains that, as more detailed profiling of gene expression and development in subcortical structures with marked sex differences emerges, it may reveal convergence of sexual differentiation and ASD risk in these populations as well (
57).
Finally, an additional factor that may contribute to the “how” of the male bias in ASD pathogenesis is that masculinization of the brain in humans, and in mammals in general, is mediated by active regulation of gene expression by SR signaling during early development, while female typical development appears to be the “default” (reviewed in
76–
78). As mentioned, the developmental epoch of such SR-controlled sexual differentiation correlates with the period most strongly implicated in ASD pathology. Male typical differentiation may therefore be more sensitive to disruptions to chromatin and gene regulation or synaptic development by ASD-associated mutations because it requires more fine-tuning during these critical windows (
70). Understanding the developmental programs that mediate sex differences may therefore offer more specific, “targetable” pathways for intervention.
Considering the Contribution of Genetics to the Development of New ASD Therapies
To date, advances in the genetics and biology of ASD has vastly outpaced efforts at clinical translation. The emerging picture of complexity with regard to both pathology and phenotype that has attended rare-variant gene discovery has tempered some of the more optimistic expectations that accompanied early successes. The notion that gene discovery would lead directly to the elaboration of therapeutic targets and traditional drug development has generally not been borne out, although for some of the highest-risk ASD mutations, the ongoing pursuit of molecular mechanisms are driving such efforts (
37,
79)
More generally, the foregoing has addressed the concept of searching for convergence, both functional and spatiotemporal, among ASD risk genes as one strategy to differentiate pleiotropic biology from “targetable” ASD pathology. Clearly this is a tall order, but the rapid pace of advance in molecular, cellular, and systems neuroscience, coupled with the increasing number of definitive molecular changes conferring ASD risk, is cause for some optimism. A driving motivation for these types of analyses is that they will allow future experiments to focus on specific mutations in specific developmental, cellular, and circuit-level contexts, which consequently can be a prelude to identifying potential therapies that are either specific to a given gene or that have an impact on multiple convergent genetic risks.
A related strategy is to focus on factors that confer resilience to large-effect genetic risks, either individually or among a set of genes demonstrating functional or spatiotemporal convergence. For example, leveraging higher-throughput model systems, our group conducted a chemical screen in zebrafish in an effort to rescue a behavioral phenotype resulting from the recapitulation of a human ASD mutation in a well-established recessive autism gene (
contactin associated protein 2) (
80). This hypothesis-free analysis found a striking enrichment in estrogenic compounds among those that rescued the phenotype. While the generalizability of this finding remains to be confirmed and the molecular mechanisms elucidated, the strategy of identifying resilience factors to large-effect genetic insults is a potentially promising avenue both to both illuminate ASD pathology and to identify novel therapeutic targets.
In contrast, one increasingly plausible alternative to elaborating “downstream” ASD pathology involves directly targeting large-effect ASD-related genetic variants as early as possible in development. With recent successes in this area in the treatment of spinal muscular atrophy (
81–
83), there is understandable interest in methods that either replace the mutated gene or manipulate gene transcription (e.g., via antisense oligonucleotides). Recall that the genomic mechanisms for many of the idiopathic ASD variants is haploinsufficiency and, consequently, that a second copy of the gene remains unaffected by the deleterious mutation. Consequently, it is plausible that relatively modest manipulations of gene expression leveraging the normal allele may be sufficient to mitigate the impact of the deleterious mutation(s). This may seem like a distant hope, but there have already been remarkable examples of personally tailored treatments for rare genetic disorders (
84) targeting gene expression and promising early results for similar strategies in “monogenic” intellectual disability syndromes (
85). In addition, the development of methods to manipulate the genome(s) of nonhuman primates may provide the critical link from more evolutionarily distant model systems experiments to human studies. All told, these advances suggest that gene targeting approaches may be closer at hand for a small fraction of individuals than is generally appreciated. And the results of these types of experiments could have a profound impact on the understanding of less invasive and more widely applicable therapeutic opportunities.
Still, the prospect for postzygotic genetic manipulation in ASD raises a host of practical and ethical challenges (
43). It is not yet clear how or whether it will be possible to deliver a therapy to the relevant location at a time when remediation is still possible. Moreover, given the wide range of outcomes associated with an individual idiopathic ASD gene, the risk-reward calculation for early intervention for many risk genes, including very complex ethical considerations, would be enormously difficult. This suggests that only the very few genes conferring the most reliable risks for the most serious developmental outcomes would be plausible candidates at the outset. In this regard, it is very likely that syndromes such as Rett, Angelman, fragile X, and tuberous sclerosis complex, will be on the vanguard of these kinds of clinical-translational efforts. For idiopathic ASD, one of the more pressing obstacles will lie in assessing and measuring change in core features of social functioning early in development and establishing experimental methods that will enable clinical trials, potentially with very small Ns. For now, it may be that associated nonsocial phenotypes, such as intellectual disability and epilepsy, will need to be the initial targets for treatment. Clearly the type of substantial investments in major collaborative efforts that transformed gene discovery a decade ago are now required to address early phenotyping and the elaboration of clinical developmental trajectories and biomarkers as an essential component of further progress on a range of novel treatment development strategies.