Social Responsiveness, an Autism Endophenotype: Genomewide Significant Linkage to Two Regions on Chromosome 8
Abstract
Objective:
Method:
Results:
Conclusions:
Method
Samples and Biomaterials
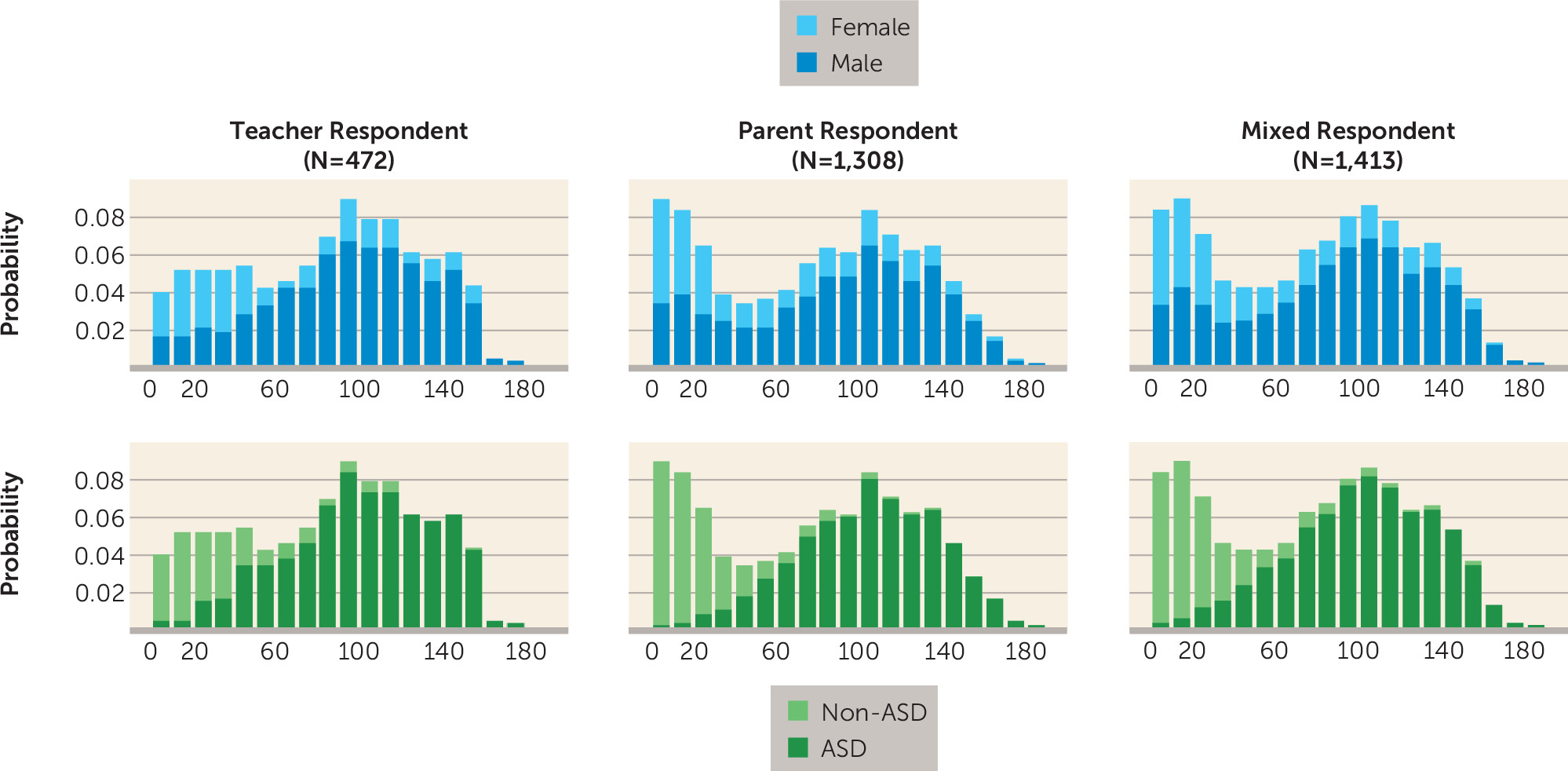
Phenotypes
SNP Genotypes
Linkage Analyses
Association Analyses
Results
Clinical/Phenotype Characteristics
| Characteristic | N | Social Responsiveness Scale Total Raw Score | |||
|---|---|---|---|---|---|
| Mean | SD | Minimum | Maximum | ||
| Families | 590 | ||||
| Phenotyped individuals | 1,480 | ||||
| Parents | |||||
| All | 67 | 36.4 | 25.3 | 5 | 113 |
| Fathers | 32 | 35.0 | 24.4 | 6 | 105 |
| Mothers | 35 | 37.7 | 26.3 | 5 | 113 |
| Autism spectrum disorder | |||||
| All | 967 | 101.8 | 33.9 | 3 | 184 |
| Males | 761 | 103.2 | 33.5 | 8 | 184 |
| Females | 206 | 96.5 | 35.1 | 3 | 169 |
| Non-autism spectrum disorder siblings | |||||
| All | 446 | 27.2 | 27.2 | 0 | 159 |
| Males | 198 | 32.2 | 32.0 | 0 | 159 |
| Females | 248 | 23.1 | 22.0 | 0 | 128 |
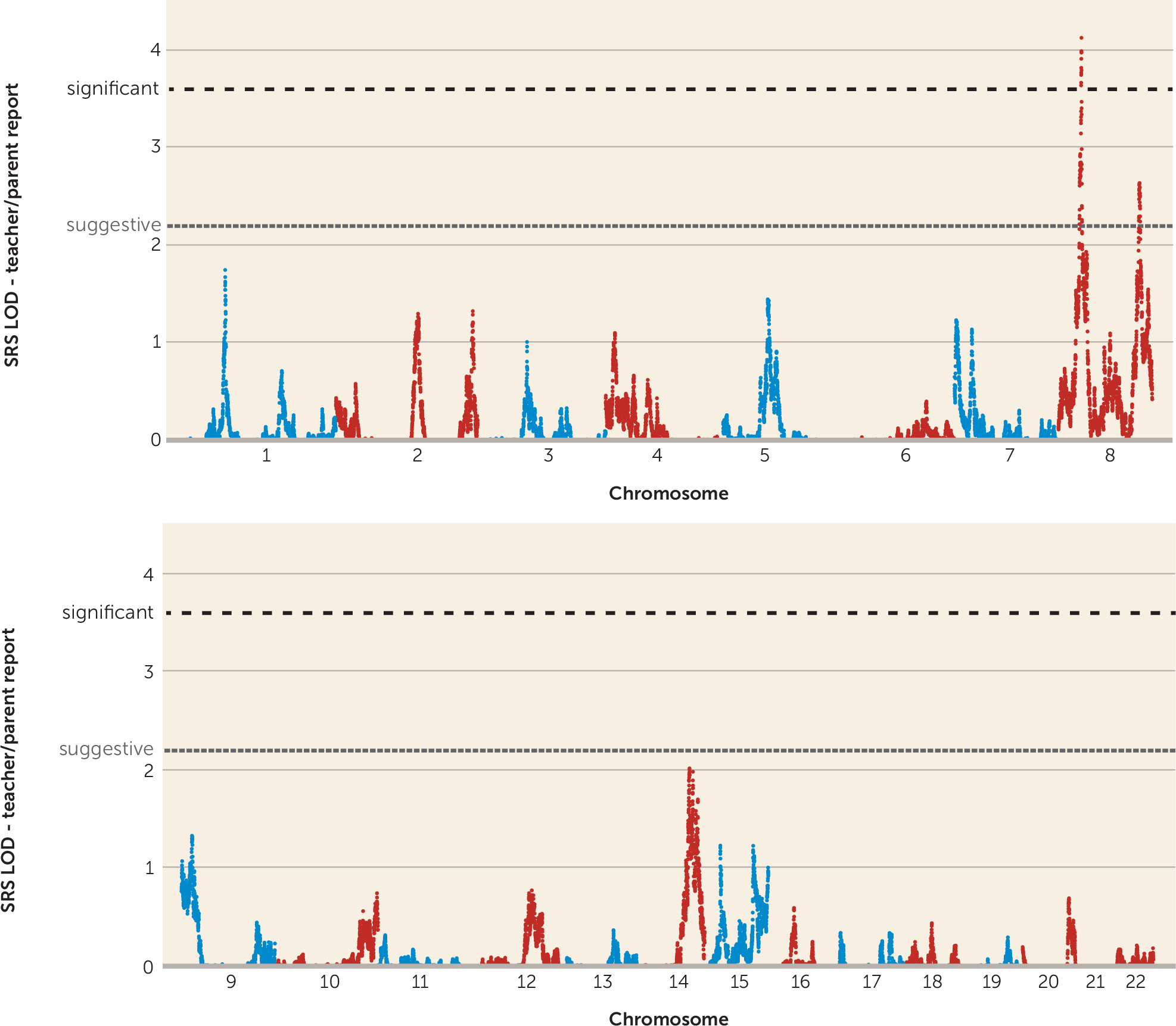
Genomewide Linkage Analysis
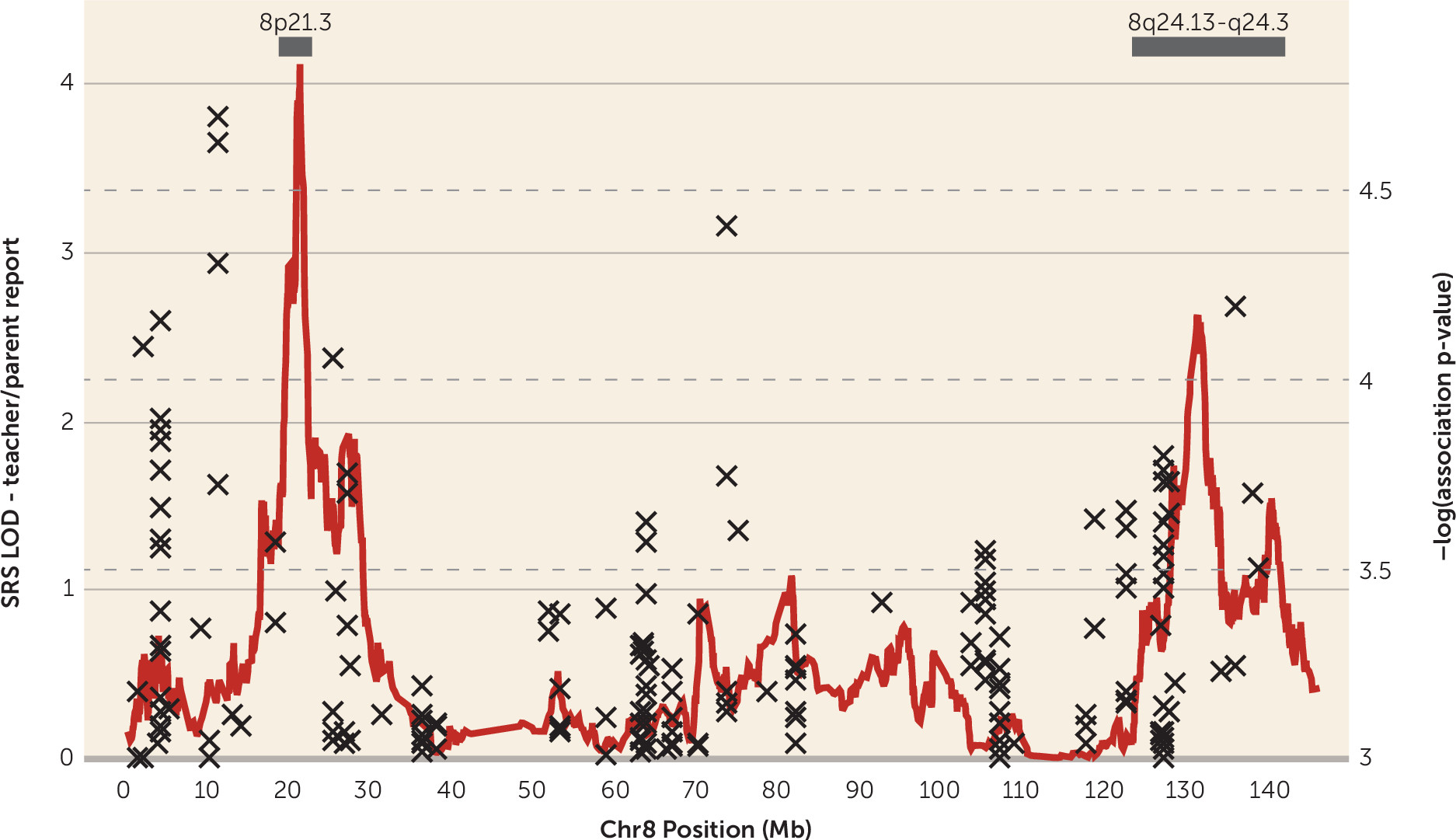
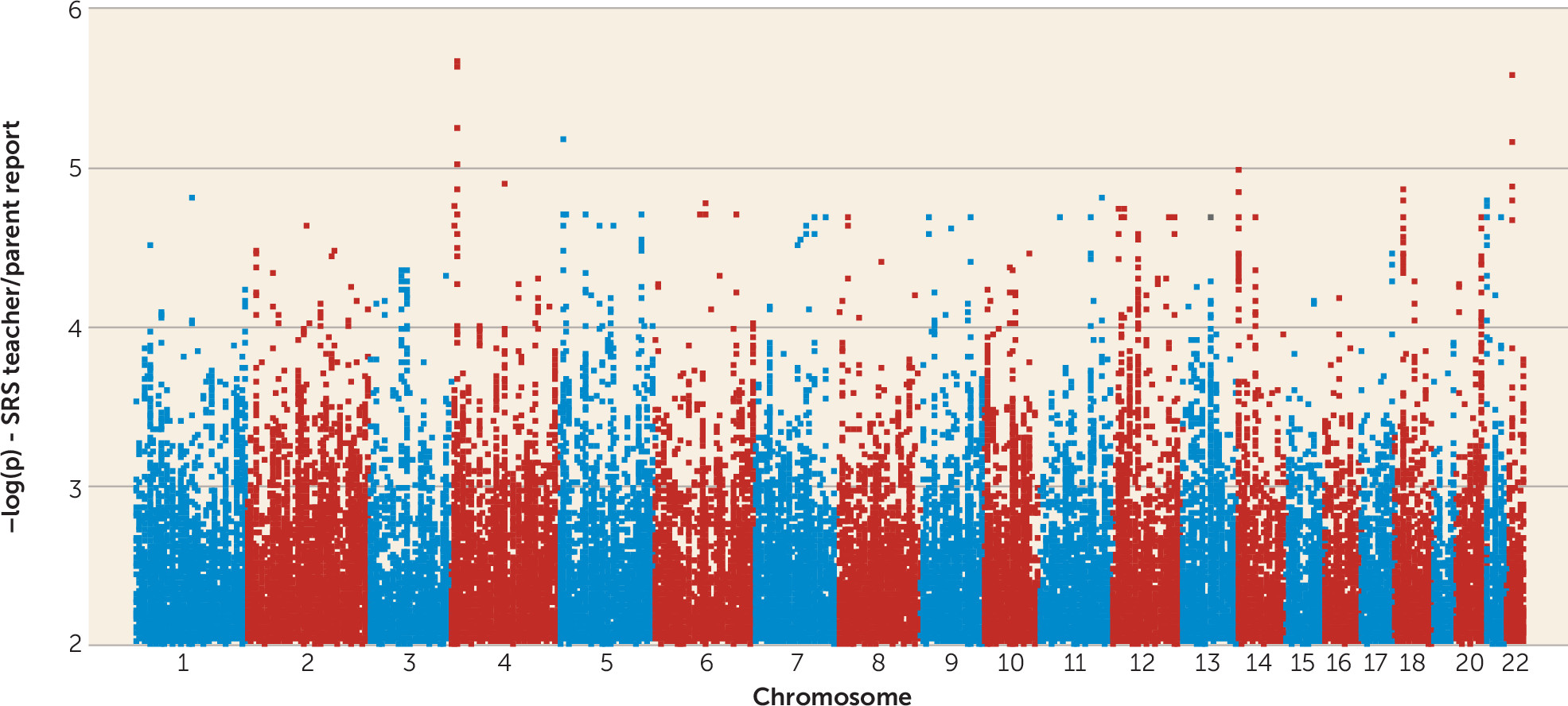
Genomewide and Linkage-Directed Association
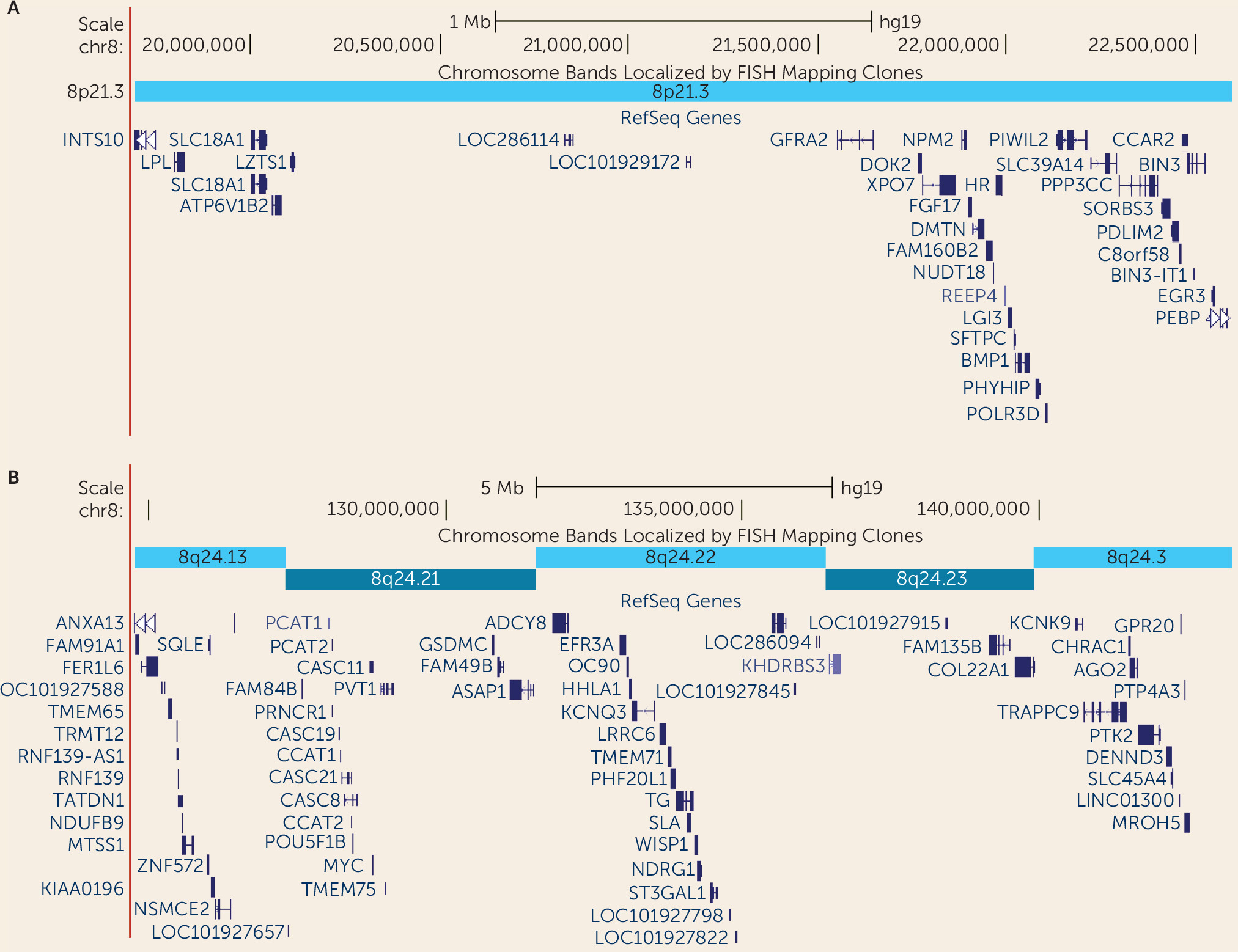
Discussion
Conclusions
Acknowledgments
Supplementary Material
- View/Download
- 3.82 MB
- View/Download
- 263.98 KB
- View/Download
- 87.36 KB
- View/Download
- 3.81 MB
References
Information & Authors
Information
Published In


History
Authors
Funding Information
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBLogin options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).