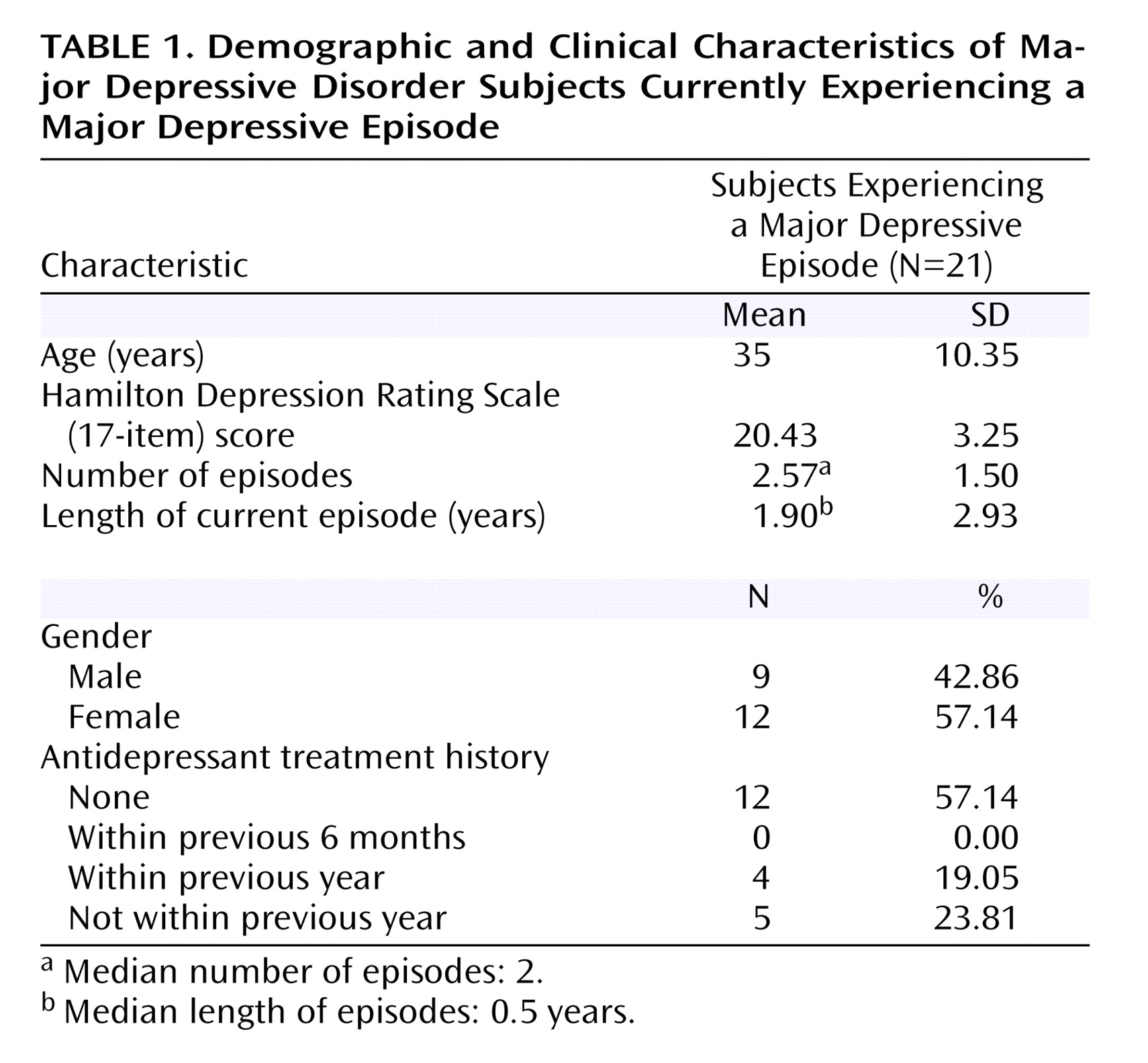
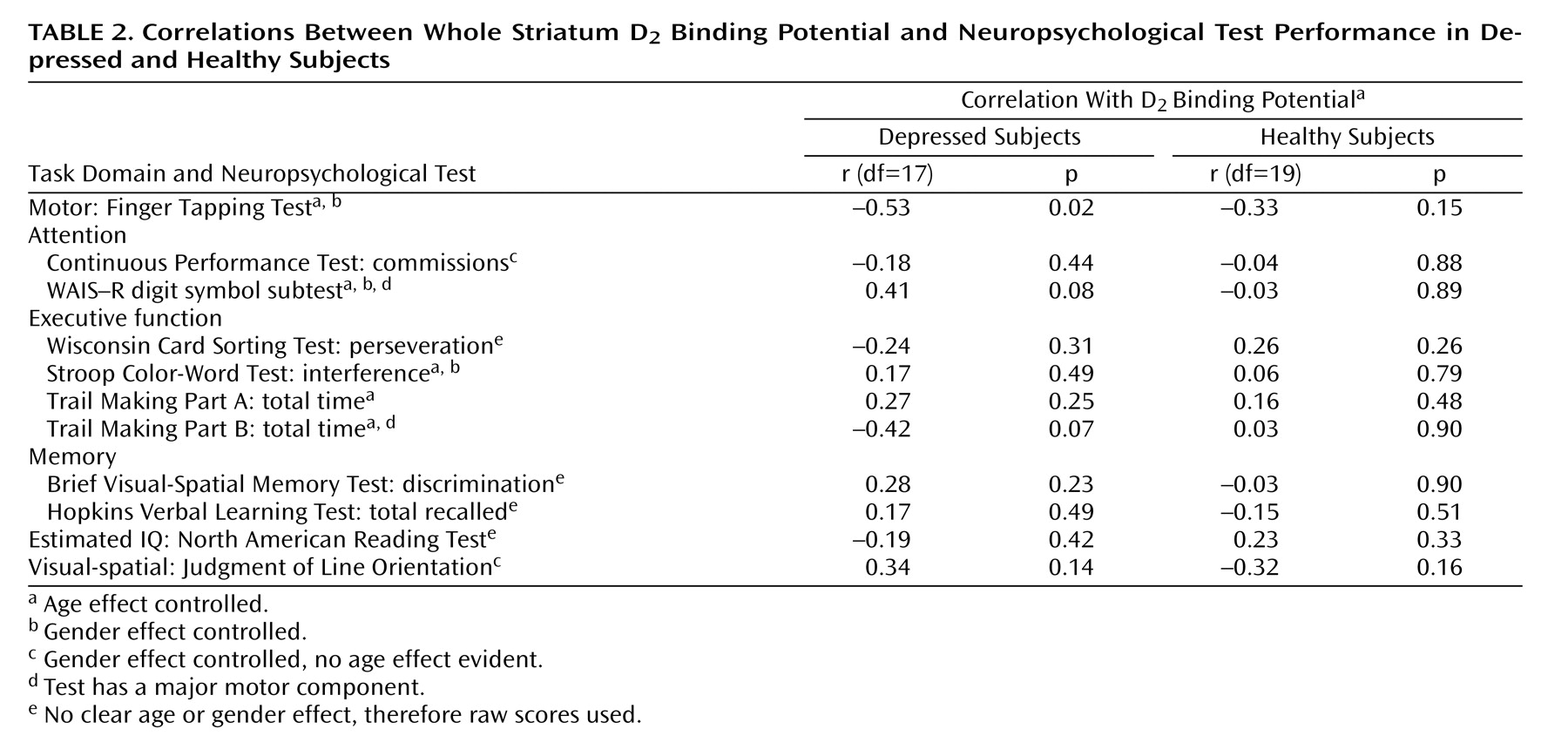
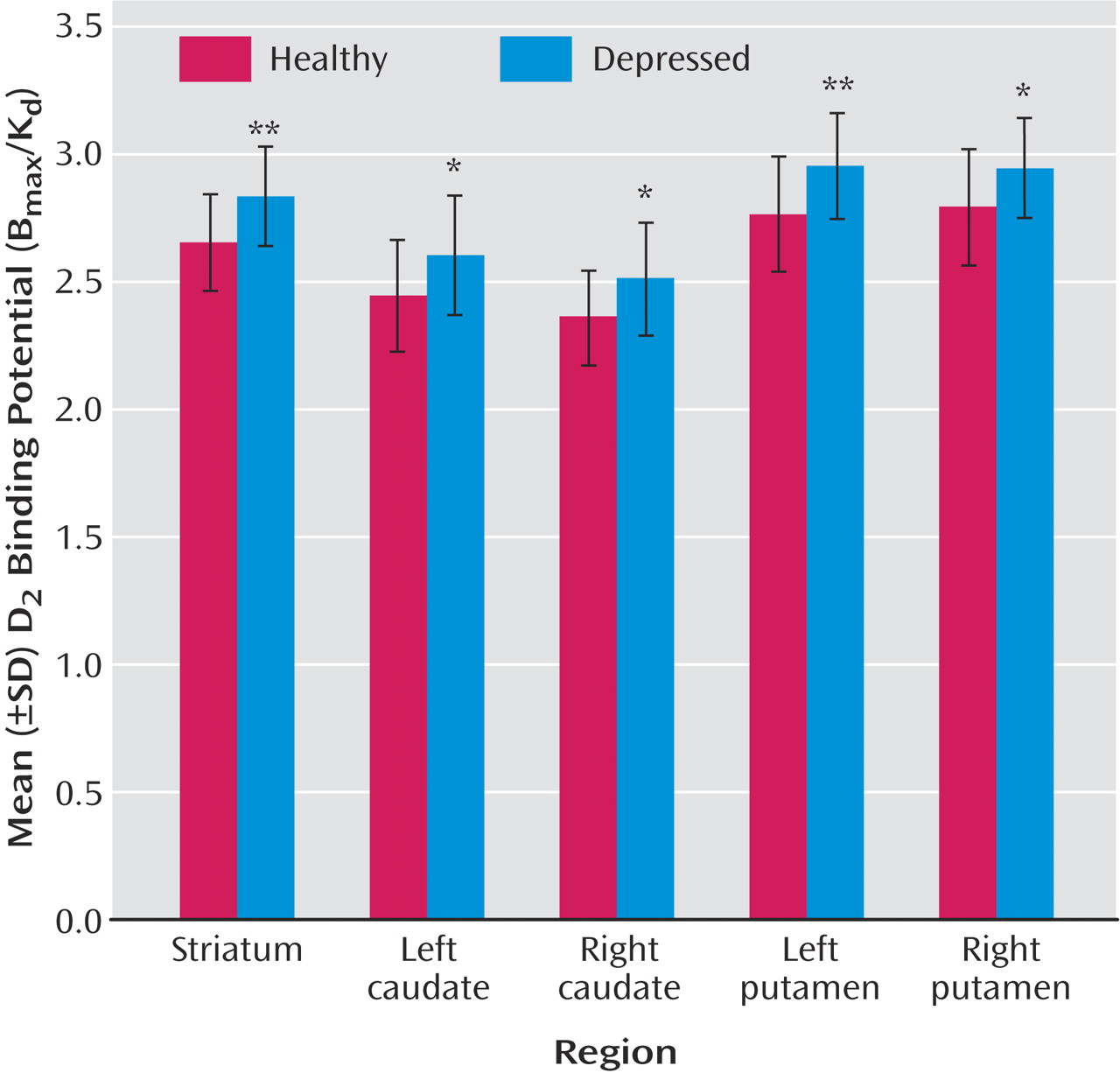
It is generally believed that pathophysiological processes implicated in major depressive disorder are reversed by antidepressant treatment. Most antidepressants raise monoamines, and the majority of these antidepressants raise serotonin and/or norepinephrine. However, a number of commonly prescribed antidepressants also have dopamine-increasing properties, the most prominent being sertraline, bupropion, and the entire class of monoamine oxidase A and A/B inhibitors
(1 –
4) . Sertraline has a moderately high affinity for the dopamine transporter
(1,
2) . Bupropion has its highest known affinity for the dopamine transporter, and the entire class of monoamine oxidase A and A/B inhibitors robustly increase striatal dopamine in animal models
(1,
3,
4) . Even though these treatments are often used, the role of dopamine during major depressive episodes is not clear.
Striatal dopamine concentrations cannot be directly measured in vivo in humans. However, [
11 C]raclopride positron emission tomography (PET) is a useful noninvasive method for investigating disease processes that may lower extracellular dopamine. [
11 C]Raclopride is a validated PET radiotracer that is selective for D
2 type receptors with a modest preference for D
2 receptors over D
3 receptors
(10 –
12) . The index of D
2 type receptors found with this method, referred to in this article as D
2 receptor binding potential, is inversely proportional to extracellular dopamine levels in animal paradigms of acute and chronic dopamine depletion as well as acute increases in dopamine
(13) . Moreover, the D
2 binding potential is elevated in human conditions associated with dopamine loss such as AMPT (alpha-methyl-paratyrosine) treatment
(14) and untreated Parkinson’s disease
(15 –
17) . The D
2 binding potential is lower after acute increases in dopamine in humans
(13) . Thus, [
11 C]raclopride PET should be useful for investigating abnormalities of striatal dopamine during major depressive episodes.
To our knowledge there have been no studies to date that have used [
11 C]raclopride PET in depressed subjects. [
123 I]IBZM single photon emission computed tomography (SPECT) is a similar method because IBZM binds to D
2 type receptors, and the D
2 binding potential found with [
123 I]IBZM SPECT shows a similar inverse relationship to extracellular dopamine levels
(13) . However, all previous investigations of striatal D
2 binding potential during major depressive episodes using [
123 I]IBZM SPECT are confounded by antidepressant use
(18 –
22) . Four
(18 –
21) of the five studies recruited subjects who had taken antidepressants within 2 weeks of scanning. This is a critical issue, since large magnitude changes in D
2 binding potential during acute administration of serotonin-increasing medications have been reported in humans
(23) . The one study that recruited some drug-free subjects
(22) cannot be considered conclusive because it did not compare the drug-free subjects to the healthy subjects. Of the five studies, only one
(20) measured both striatal D
2 binding potential and motor speed with neuropsychological testing. This study is also not definitive because most of the subjects were taking antidepressant medication, subjects with bipolar disorder were included, and the effects of age were not addressed. Striatal D
2 binding potential declines with age
(24), and motor speed declines with age
(25) . Therefore, controlling for the effect of age on both variables is essential or else a spurious association between the two is possible.
The underlying hypothetical model for the present study was that extracellular dopamine would be low in the putamen during major depressive episodes when motor retardation is present. The evidence supporting this model is that CSF concentration of the dopamine metabolite, HVA, is low during depression with motor retardation
(8,
9) and that low dopamine neurotransmission in the putamen is associated with motor retardation in several other diseases
(5,
6) . Extracellular striatal dopamine cannot be measured directly in vivo in humans; however, the D
2 binding potential found with [
11 C]raclopride PET is inversely correlated with extracellular dopamine levels
(13,
14,
16) . Therefore, our specific hypothesis was that putamen D
2 binding potential would be elevated in depressed subjects exhibiting motor retardation.
Discussion
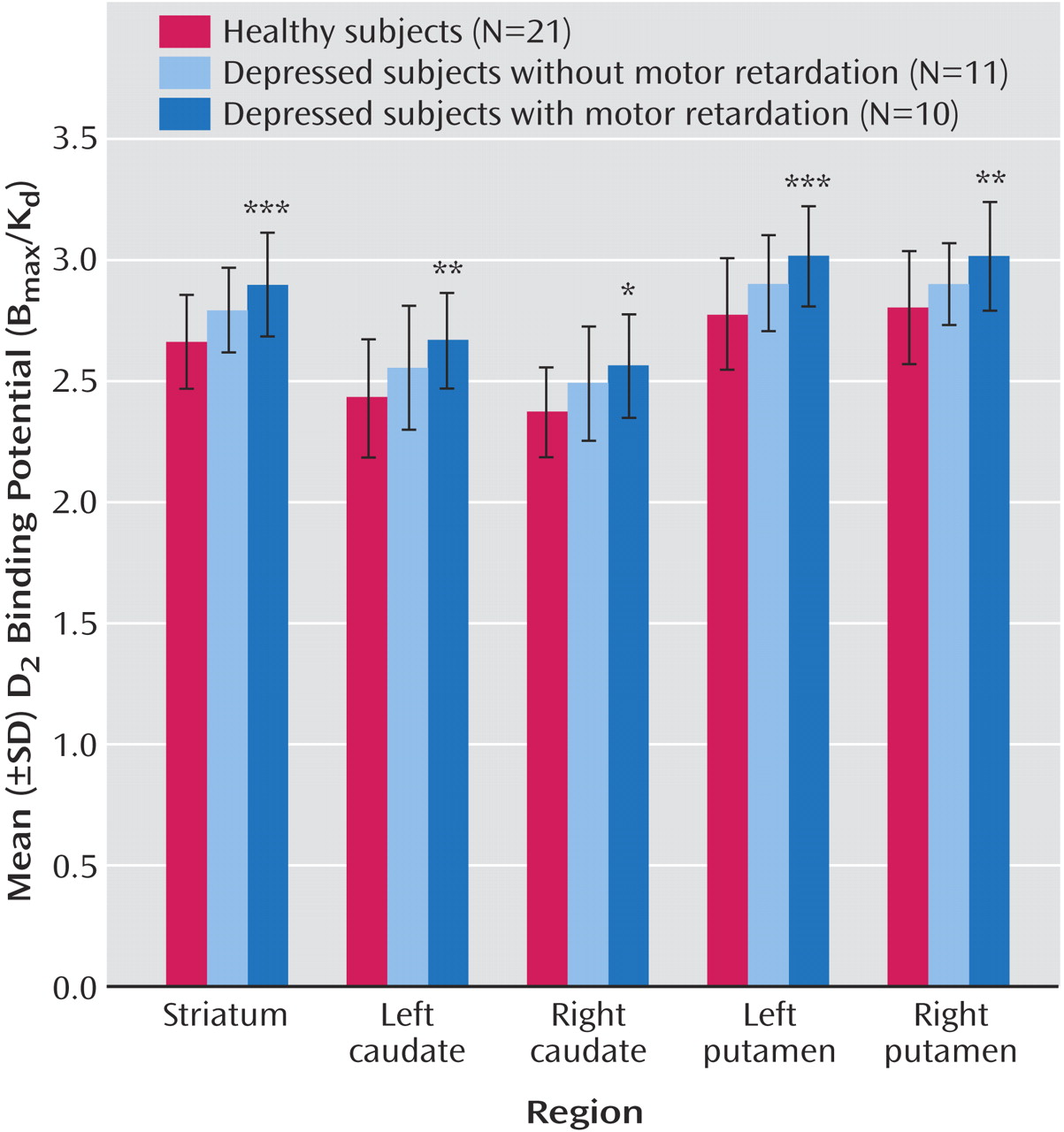
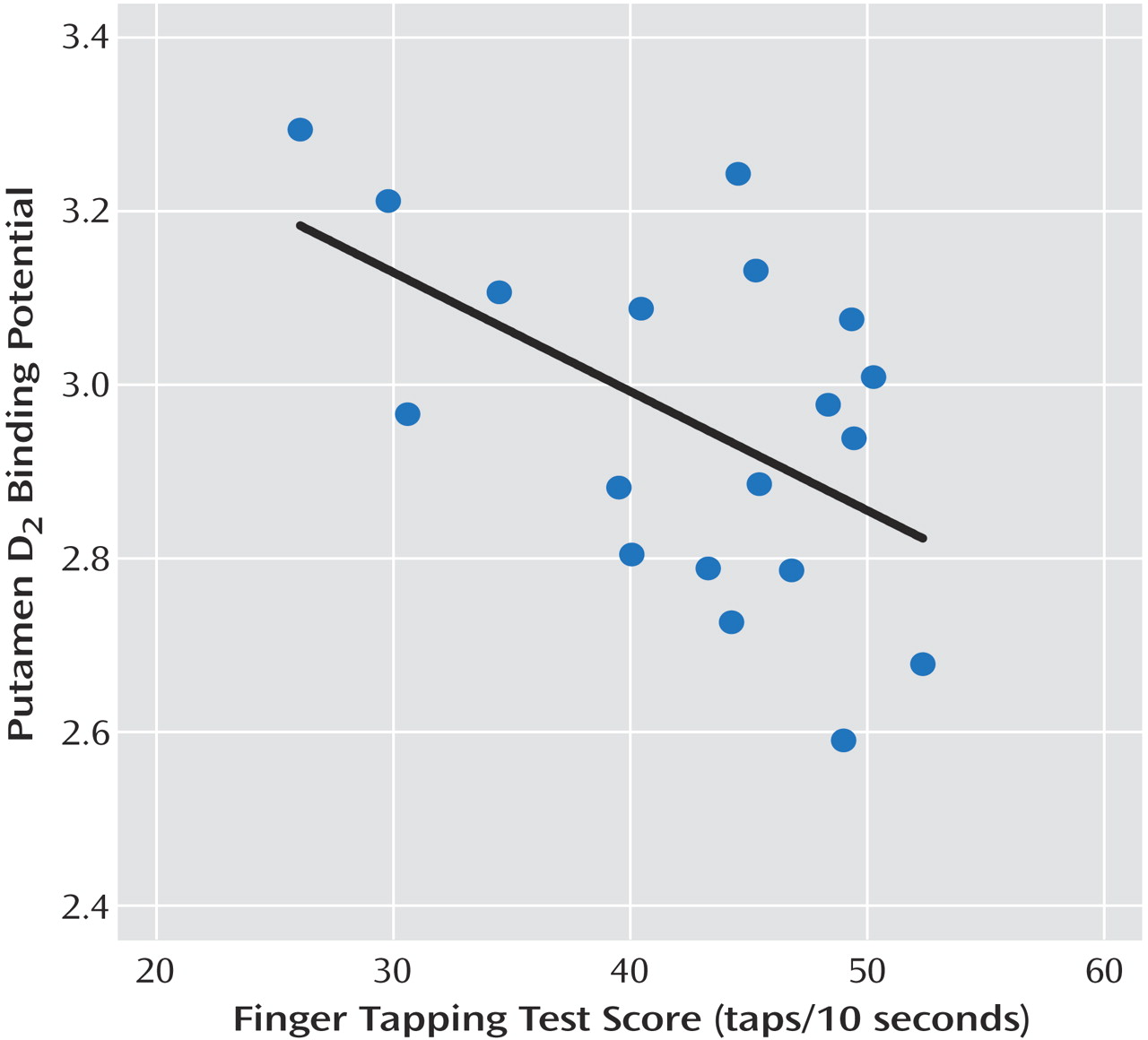
This is the first study to investigate striatal D 2 binding potential in medication-free, nonsmoking depressed subjects. The main findings were that caudate and putamen D 2 binding potential were elevated in the depressed group as compared with the healthy group, and that putamen D 2 binding potential was most significantly elevated in the depressed subgroup with motor retardation. Moreover, there was a significant correlation between putamen D 2 binding potential and motor speed in the depressed group. These findings are important for understanding the pathophysiology of depression because they represent a striatal dopaminergic disturbance during depression that is most prominent when motor retardation is present. This has implications for the monoamine theory of depression. In addition, these findings have important implications for treatment, since this dopamine abnormality may be targeted by dopamine-increasing antidepressants.
The best interpretation of these findings is that putamen dopamine is low in depression with motor retardation. Elevations in D
2 binding potential measured with [
11 C]raclopride PET may occur when dopamine is low
(13,
14), and the most significant elevations in putamen D
2 binding potential were observed in the depressed subjects with motor retardation (
Figure 3 ). These results are consistent with previous reports of lower CSF metabolite of dopamine in depressed subjects with motor retardation
(8,
9) and the general disease model associating motor retardation with impaired dopaminergic neurotransmission in the putamen
(5,
6) . These results are also consistent with our previous finding of lower dopamine transporter binding potential in a similar group of medication-free depressed subjects
(34), since dopamine transporter density can decrease during chronic dopamine-depleting paradigms
(36,
37) . The other investigation of striatal dopamine transporter in medication-free depressed subjects sampled those with seasonal affective disorder and also found lower striatal dopamine transporter binding potential
(38) .
The results of the present study have important implications for understanding the neurochemistry of major depressive disorder. In many versions of the monoamine theory of depression, major depressive disorder is viewed as having a pathology of homogenous monoamine loss. A contrasting view is that specific monoamines are lowest when specific symptoms of depression are most severe
(35) . Although putamen D
2 binding potential was elevated in the depressed group as compared with the healthy group, the data in
Figure 3 show that the elevation in putamen (and striatal) D
2 binding potential was driven by the depressed group with motor retardation. Since putamen D
2 binding potential is highest in the depressed subjects with motor retardation and since measurement of D
2 binding potential with [
11 C]raclopride PET is inversely correlated with extracellular dopamine levels
(13,
14,
16), the results argue for heterogeneous monoamine loss in depression (with greater specific monoamine loss when specific symptoms are more severe).
The interpretation that extracellular dopamine is low in depressed subjects with motor retardation has substantial implications for treatment because it argues for a preferential response in this subtype to antidepressants that induce sustained dopamine agonist effects such as dopamine reuptake inhibitors (i.e., bupropion and sertraline) and monoamine oxidase A and A/B inhibitors
(1 –
4) . Moreover, it is likely that further dopamine reuptake inhibitor development will occur, since current treatments are unlikely to be optimally therapeutic at this target site. For example, in contrast to the 80% or greater occupancy of serotonin transporters typically found at therapeutic dosing of serotonin reuptake inhibitors
(35), dopamine transporter blockade with sustained-release bupropion has been estimated between 0% and 26%
(39 –
41) . The other commonly used antidepressant with dopamine reuptake inhibitor properties, sertraline, has approximately two orders of magnitude higher affinity for the serotonin transporter over the dopamine transporter
(1,
2), and it is probable that a closer affinity ratio between these two sites would be more optimal for therapeutic effect. A related class, dopamine-releasing medications, is available, but their mechanism is dependent upon intact dopamine storage
(42), making them less clinically relevant. It is well known that serotonin-releasing medications like
d -fenfluramine are not therapeutically relevant for treating depression. It is also theoretically possible that dopamine agonists that stimulate dopamine receptors in a similar manner to dopamine itself could also prove to be therapeutically useful in treating motor retardation during major depression. The results of the present study argue for 1) the use of existing dopamine-increasing medications to treat depression with motor retardation and 2) the development of antidepressants with higher dopamine reuptake inhibitor blockade for depression with motor retardation.
Striatal D
2 binding potential was 9% higher in the putamen of depressed subjects with motor retardation as compared with the healthy subjects. Although this elevation of putamen D
2 binding potential in depressed subjects with motor retardation was significantly higher than healthy subjects, it is less than what occurs in early untreated Parkinson’s disease (estimates with [
11 C]raclopride PET measurement typically range from 7% to 30% [
15 –
17 ], averaging at 20%) or after a severe acute dopamine depletion with alpha-methyl-paratyrosine (18%
[14] ). This is not surprising: the loss of dopamine neurons in Parkinson’s disease is generally considered a large magnitude effect, and the version of the alpha-methyl-paratyrosine challenge used by Verhoeff et al.
(14) was a drastic paradigm tolerated for only a brief period under inpatient supervision.
This study has limitations inherent in the measurement of D
2 binding potential that are common with in vivo imaging studies of disease states. Striatal D
2 binding potential as measured with [
11 C]raclopride reflects a combination of D
2 receptor density, affinity, and accessibility of the radioligand to D
2 receptor sites. While D
2 binding potential can certainly increase under conditions of acute and chronic dopamine depletion
(13,
14), there are a number of other potential explanations for a greater striatal D
2 binding potential. Therefore, our interpretation of low extracellular dopamine is also dependent upon other dopamine-related investigations in major depressive disorder. We did not measure hedonia, since neuropsychological measurements of hedonia need to be developed for major depressive disorder. Self-report of hedonia is potentially prone to biases of severely negativistic thinking (where every experience is viewed pessimistically, resulting in underreporting) and anxiety (where people are hesitant to incorrectly report and therefore underreport). Some PET study designs also measure D
2 binding potential after dopamine depletion and assume that the dopamine depletion would result in similar final dopamine levels in patients and healthy comparison subjects
(13) . This assumption seems contradictory to our hypothesized model of abnormally regulated extracellular dopamine levels in depression, so we did not apply a dopamine-depleting paradigm. Our previous work found that the striatal dopamine transporter binding potential is lower in depression
(34) —as would be expected during a dopamine-lowering process
(36,
37) —and when it is lowest, it is actually protective against motor retardation. We surmised that a lower striatal dopamine transporter binding potential would result in less clearance of extracellular dopamine and would be protective against a different dopamine-lowering process. Promising mechanisms for a major dopamine lowering process include dysregulations of enzymes of dopamine synthesis or dopamine metabolism, and these will be studied in future research.
This study was the first investigation of striatal D
2 binding potential in drug-free, nonsmoking, currently depressed subjects with a reasonably large patient group size. We found that depressed subjects with motor retardation had a significantly elevated striatal D
2 binding potential relative to healthy volunteers. Moreover, among depressed subjects, higher striatal D
2 binding potential was correlated with more severe motor retardation. The best explanation for these findings is that lower extracellular dopamine occurs in depressed subjects with motor retardation, since this explanation is consistent with reports of low CSF HVA in depression with motor retardation
(8,
9), low dopamine transporter binding potential in depression
(34), and an association between putamen dopaminergic pathology and motor retardation in other disease models
(5,
6,
15) . Low dopamine pathology in depression with motor retardation has important implications for the treatment of motor retardation in depression. This subtype of depression should benefit from dopamine-increasing medications, and future clinical trials of dopamine reuptake inhibitor treatments should be conducted in depression with motor retardation.