Significant life stress, including early life stress, represents a major risk factor for development of major depression
(1) . Once manifested, major depression has been associated with enhanced tonic activation of the innate immune system, including increased plasma proinflammatory cytokines such as interleukin (IL)-6
(2) . In nondepressed individuals, exposure to psychosocial stress has been associated with increased plasma IL-6 responses
(3), as well as activation of nuclear factor (NF)-kB
(4), a transcription factor that serves as a lynchpin in the inflammatory signaling cascade. Whether stress-induced increases in inflammatory activity are exaggerated in patients with psychiatric disease has not been examined. Given the relationship between early life stress and depression and the fact that psychosocial stress activates innate immune responses, we hypothesized that patients with major depression and increased early life stress would exhibit enhanced responsiveness of plasma IL-6 and NF-κB to psychosocial challenge.
Method
Twenty-eight male subjects, medically healthy as determined by physical examination and laboratory testing, were recruited between May 2003 and April 2005 as part of a study investigating the neurobiology of early life stress and major depression. Fourteen patients had current major depression, determined by Structured Clinical Interview for DSM-IV (SCID) (mean age=29.9 years [SD=9.6], mean body mass index [BMI]=27.8 [SD=7.1]). The other 14 participants were nondepressed according to the SCID, 21-item Hamilton depression scale (score ≤8), and Zung scale (score <50) (mean age=29.4 years [SD=8.0], mean BMI=24.3 [SD=2.6]). Mean Hamilton depression scale scores were 20.7 (SD=5.5) for major depression patients and 3.6 (SD=2.6) for healthy comparison subjects (t=10.62, df=26, p<0.001). Early life stress experiences were quantified using the Childhood Trauma Questionnaire
(5) . Mean Childhood Trauma Questionnaire total scores were significantly higher (p<0.001) in depressed (69.0 [SD=25.08]) versus comparison participants (35.0 [SD=15.68]). Past or current psychotic symptoms, bipolar disorder, substance abuse/dependence within 1 year, current eating disorder, and use of psychotropic medication were exclusionary. All participants provided written informed consent. The study was approved by the Emory University Institutional Review Board.
After an overnight stay in the Emory General Clinical Research Center, participants underwent the Trier Social Stress Test at 3 p.m. as previously described
(4) . Ten minutes of preparation/anticipation were followed by a public speaking task (5 minutes) and a mental arithmetic task (5 minutes). Blood (7 ml) was collected from an indwelling venous catheter into chilled EDTA-coated monovettes and centrifuged immediately. Plasma was stored at –80°C. The remaining cell pellet was reconstituted with 1x PBS and loaded onto a Ficoll gradient to isolate PBMCs, which were washed twice in cold 1x PBS before storage at –80°C in fetal bovine serum/10%DMSO.
Plasma IL-6 concentrations were measured by enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, Minn.). NF-κB pathway activation was measured in a subset of depressed patients (n=9) and comparison subjects (n=12). On the basis of a report that NF-κB activation peaks approximately 30 minutes after Trier Social Stress Test initiation
(4), relative PBMC NF-κB DNA binding before and 30 minutes after the Trier Social Stress Test was compared. Absolute baseline nuclear NF-κB DNA binding was measured against a recombinant p65 standard curve (Active Motif, Carlsbad, Calif.). Nuclear extracts were obtained from PBMCs (5–7×10
6 cells/extraction) using a low/high salt extraction kit (Pierce Biotechnology, Rockford, Ill.). Binding of NF-κB to its consensus DNA sequence was measured using DNA-binding ELISA (Active Motif). Lymphocyte subsets within isolated PBMCs (1×10
6 cells obtained concurrently with NF-κB measures) were enumerated using fluorochrome-conjugated antibodies (BD Biosciences, Franklin Lakes, N.J.) directed toward CD16/56 (NK cells), CD4 (class II major histocompatibility complex [MHC] T cells), CD8 (class I MHC T cells), and CD20 (B cells) in a subset of depressed patients (n=7) and comparison subjects (n=10). Cells were analyzed using a FACScan flow cytometer (Beckham Coulter, Fullerton, Calif.) and FlowJo software.
Plasma IL-6 concentrations were compared between groups using 2-way ANOVA for repeated measures (time x group), with Huynh-Feldt adjustment for unequal variance. Differences from lowest to highest IL-6 concentrations over the time course (ΔIL-6) were compared between groups by Mann-Whitney-U test. Independent sample t tests were used for between-group comparisons of baseline NF-κB activation and change in NF-κB DNA-binding and NK cell percentage from baseline to 30 minutes after Trier Social Stress Test onset (ΔNF-κB and ΔNK, respectively). Spearman correlation coefficients were calculated to examine the association between relevant continuous clinical and biological variables. Multiple regression analyses were used to examine the independent contribution of the Hamilton depression scale and Childhood Trauma Questionnaire scores to ΔIL-6 and ΔNF-κB. ΔIL-6 was log transformed for the purposes of these analyses. Tests of significance were two-sided with an alpha level of p≤0.05.
Results
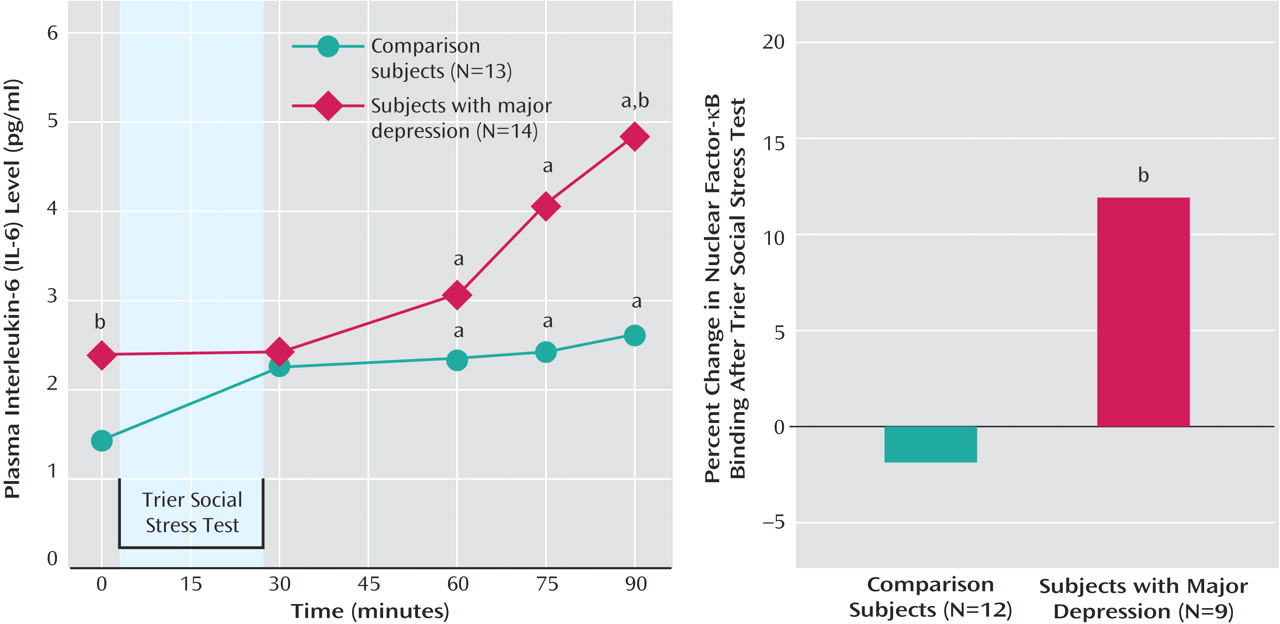
All participants displayed an increase in plasma IL-6 concentrations during Trier Social Stress Test challenge over time (F=10.32, df=1.92, 48.10, p<0.001). There was also a significant interaction between sampling time and group (F=3.29, df=1.92, 48.10, p<0.05). Post-hoc analysis (Bonferroni corrected for multiple comparisons) indicated that both groups had greater plasma IL-6 concentrations at 60, 75, and 90 minutes after Trier Social Stress Test onset relative to baseline. Depressed patients had higher IL-6 concentrations than comparison subjects at baseline and 90 minutes poststressor (
Figure 1 ). Depressed patients also displayed a greater IL-6 response (ΔIL-6) to the Trier Social Stress Test than comparison subjects (Mann-Whitney U=44, p<0.05).
Baseline NF-κB DNA-binding (p65) did not differ between comparison subjects (4.8 ng [SD=1.7]) and depressed patients (5.2 ng [SD=2.5]). However, ΔNF-κB was greater in depressed patients (t=2.10, df=19, p<0.05) (
Figure 1 ).
Hamilton depression scale scores were significantly correlated with ΔIL-6 (r s =0.44, N=27, p=0.02) and ΔNF-κB (r s =0.46, N=21, p<0.04). No significant correlations were observed between ΔIL-6 and ΔNF-κB or between Childhood Trauma Questionnaire scores and inflammatory measures. Multiple regression analyses indicated that Hamilton depression scale score was a significant independent predictor of ΔIL-6 (r p =0.42, p<0.04) and ΔNF-κB (r p =0.45, p<0.05, respectively), whereas partial correlation coefficients between Childhood Trauma Questionnaire score and immune variables (when controlling for Hamilton depression scale score) were not significant.
Among the lymphocyte subsets, only NK cell percentage significantly increased in response to Trier Social Stress Test in both groups. However, ΔNK did not differ between major depression patients (15.1% [SD=7.9]) and comparison subjects (14.8% [SD=8.6]). Moreover, ΔNF-κB and ΔNK were not correlated (r s =0.015, N=14, p=0.96), nor was ΔNK correlated with ΔIL-6 (r s =0.050, N=16, p=0.85) or Hamilton depression scale score (r s =0.28, N=17, p=0.27). In addition, ΔNF-κB was not correlated with Trier Social Stress Test-induced changes in other PBMC subsets (data not shown).
Discussion
Although previous studies have shown evidence of increased baseline activation of the inflammatory response in major depression patients (e.g.,
2 ), the current study extends these findings by providing the first evidence that the inflammatory response to stress is exaggerated in major depression patients, specifically male patients with major depression and increased early life stress. This difference was observable only at later timepoints following Trier Social Stress Test onset, and may explain why previous studies have failed to identify increased inflammatory responses after psychological stress
(6) . Interestingly, baseline elevations in plasma IL-6 concentrations in major depression patients with increased early life stress were similar to maximal stress-induced IL-6 responses in comparison subjects, suggesting that male major depression patients with increased early life stress may chronically exhibit stressed-induced increases in inflammatory markers that are further exacerbated by exposure to acute stress. These findings indicate that depressed patients with increased early life stress exhibit both a baseline hyperinflammatory state coupled with a hyper-responsive inflammatory response to stress, which together may contribute to medical comorbidities associated with major depression and inflammation such as cardiovascular disease
(7) .
Previous work suggests that stress-induced changes in NF-κB DNA binding may result from shifts in lymphocyte subpopulations during exercise stress
(8) . In particular, NK cells have been shown to exhibit high constitutive levels of NF-κB DNA binding, and NK cell percentage increases following stress. While NK cell percentages did increase as a result of the Trier Social Stress Test, the magnitude of this increase did not differ between groups, nor did it correlate with stress-induced changes in NF-κB or IL-6. The lack of a relationship between NK cells and IL-6 is consistent with a recent report
(9) . However, dissociation between NF-κB and NK cell percentage in this and previous studies may be a function of the stressors employed (public speaking/mental arithmetic), which qualitatively differ from exercise.
Of particular importance is that major depression patients in this study had significantly higher Childhood Trauma Questionnaire scores (early life stress) than comparison subjects. As previously reported, early life stress is commonly associated with major depression
(10) and has been associated with distinct neurobiological responses to stress
(11) . Indeed, previous stressor exposure has been shown to sensitize subsequent immune responses to immune challenge
(12), and therefore the findings may result from an interaction between major depression and early life stress. Nevertheless, depression scores exhibited a significant independent relationship with immune variables, whereas Childhood Trauma Questionnaire scores did not. However, due to the relatively small study group size and study design, the relative contribution of early life stress alone or in combination with major depression cannot be definitively established. Correlation coefficients observed between Childhood Trauma Questionnaire scores and inflammatory markers represented small-to-medium effect sizes that would require total sample sizes of N≥70 to have sufficient power (80%) to detect significance using a two-sided test (p≤0.05)
(13) . These potential relationships would be best revealed in future studies using a stratified design with a larger sample size.
The mechanisms of the observed effects may relate to changes in neuroendocrine function, including increased sympathetic nervous system responses and/or altered glucocorticoid feedback regulation. Sympathetic nervous system activation has been shown to enhance inflammatory responses
(4), and major depression patients with early life stress have been shown to exhibit enhanced sympathetic nervous system responses to stressor challenge
(11) . In addition, alterations in glucocorticoid feedback, which play an important role in modulating inflammatory responses, have been described in major depression patients
(14) . Taken together, the data suggest that further studies examining the independent contributions of major depression and early life stress to the inflammatory response to stress as well as the mechanisms involved are warranted.