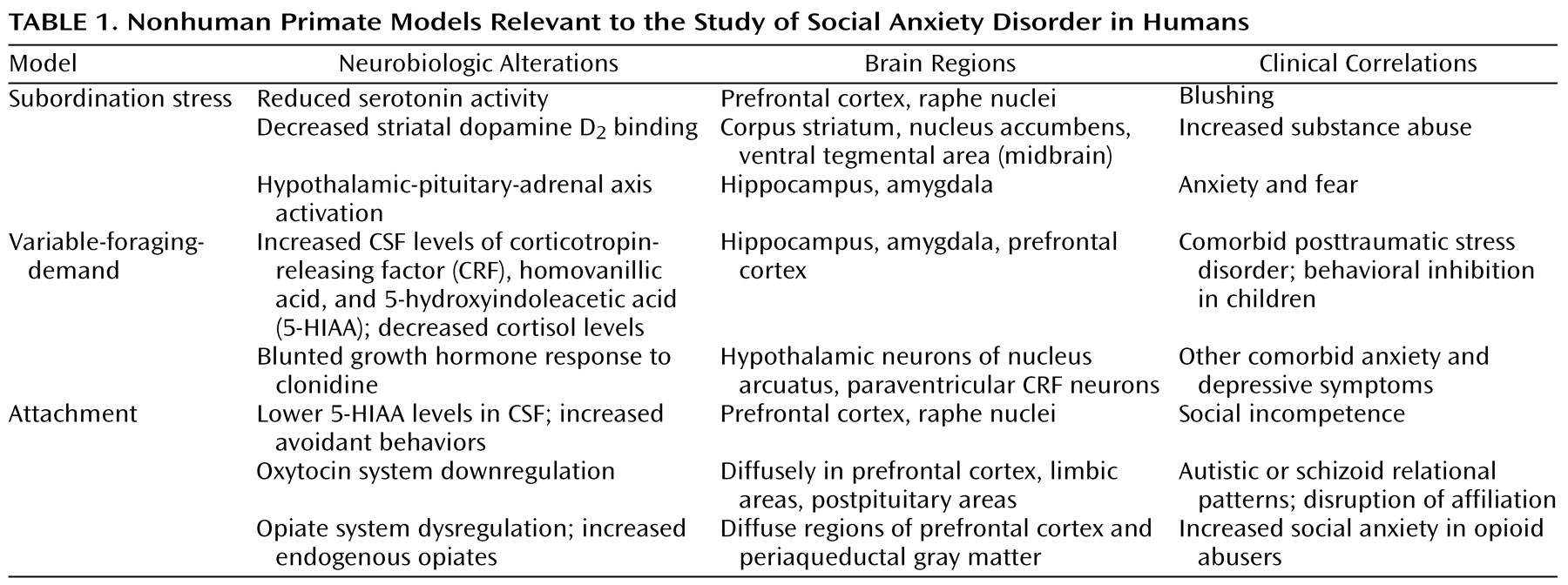
Social anxiety disorder, also known as social phobia, is a common and disabling psychiatric illness that is characterized by an excessive fear and/or avoidance of situations in which an individual feels scrutinized by others and is fearful of a negative evaluation by others. Although it is the most common of the DSM-IV anxiety disorders, there is a dearth of clinical neurobiological research on social anxiety disorder and few preclinical models. This review focuses on the generalized subtype, which involves the fear of a wide range of social situations, with the goal of proposing several neurobiological mechanisms that may account for the symptoms of this disorder. We begin with an overview of three nonhuman primate models that are particularly relevant to social anxiety. Next, we review recent literature in the clinical neurobiology of social anxiety disorder, focusing on important findings in developmental neurobiology and genetics. Our findings suggest that social anxiety disorder should be reconceptualized as a chronic neurodevelopmental illness instead of an episodic de novo adult disorder, a semantic distinction with important treatment implications.
Genetics of Social Anxiety Disorder
Although there is increasing evidence that social anxiety disorder and its childhood variants, including behavioral inhibition and shyness, have a strong familial basis, the genetics of the disorder have not been adequately studied. Several early studies
(58,
59) established a familial link, but only for the generalized subtype
(59). It was reported that if a proband has a diagnosis of social anxiety disorder, the percentage of first-degree relatives with the illness was 15%, which was greater than the 10% finding in subjects with agoraphobia and less than the 31% seen in subjects with simple phobia
(49). Subsequently, a larger study
(60) showed that the generalized subtype was markedly increased in frequency (approximately 10 times greater) among first-degree relatives of generalized social phobic probands. Another study
(61) demonstrated that the children of patients with social anxiety disorder were at an increased risk of developing this disorder and other anxiety disorders.
The low genetic concordance rates for social anxiety disorder in monozygotic twins
(62) have suggested that genetics plays a limited role in its development. As we suggested for panic disorder
(1), what appears to be inherited is a susceptibility to social anxiety, not the disorder itself. Although no systematic genetic linkage studies employing a genomic scan or search among candidate genes have been conducted for social anxiety disorder so far, such studies are underway for panic disorder
(63) and OCD
(64). Likewise, molecular genetic studies of candidate genes for the several neurotransmitter systems implicated in social anxiety, notably the serotonin transporter and dopamine receptor and their various subtypes, have allowed for associations between specific genes and behavioral traits, such as harm avoidance and novelty seeking
(65,
66)—characteristics relevant to the social anxiety disorder phenotype. Thus, genetic and family studies in social anxiety disorder are still in their infancy but support longitudinal clinical data that are suggestive of links between childhood and adult variants of the disorder.
Neuroimaging
Neuroimaging studies to date have primarily focused on basal ganglia or striatal pathology and have shown preliminary evidence of impaired dopaminergic functioning in these regions. The interest in these specific brain regions followed accumulating clinically based evidence of dopaminergic deficits in social anxiety disorder (
Appendix 1). Neuroanatomically, of the four major dopamine pathways in the CNS, dysfunctions of the mesocortical and mesolimbic (ventral striatal, including the nucleus accumbens) pathways appear most relevant to social anxiety, with a presumed lesser importance of tuberoinfundibular and nigrostriatal (dorsostriatal) pathways, although published imaging studies do not provide sufficient spatial resolution to make this determination.
A study by Tiihonen et al.
(82) reported a decrease in striatal dopamine reuptake sites on SPECT in patients with social anxiety disorder compared to normal volunteers, which suggests a deficit of dopaminergic innervation into the striatum. The authors suggested that the lowered dopamine reuptake site density reflects an overall smaller number of dopaminergic synapses and neurons in the striatum of patients with social anxiety disorder. The recent [
123I]iodobenzamide ([
123I]IBZM) SPECT study of Schneier et al.
(5), which showed reduced mean D
2 receptor binding in the striatum, implicated dopaminergic hypofunction in the striatum. However, the interpretation of this report is difficult to reconcile with the report by Tiihonen et al. of decreased dopamine transporter binding, in that decreased binding potentials of the SPECT radiotracer [
123I]IBZM could also reflect
increased levels of free dopamine in the vicinity of D
2 receptors, altered affinity of D
2 receptors for dopamine, or some combination of these factors. It was recently argued that SPECT or PET studies measuring dopamine binding after changes in synaptic dopamine levels are probably more complex than accounted for by simple binding occupancy models and might involve changes in the subcellular distribution of receptors
(83). Indeed, most of the variance in D
2 receptor binding appears to be due to alterations in receptor expression, whereas endogenous dopamine levels contribute to only about 10%–20% of the variance (personal communication, Marc Laruelle, M.D., 2001).
Most neuroimaging studies not specifically focusing on dopamine systems have detected basal ganglia and cortical abnormalities, and one study suggested amygdala involvement. Using magnetic resonance spectroscopy (MRS), Davidson et al.
(84) reported a decrease in choline and creatine signal-to-noise ratios in the subcortical, thalamic, and caudate areas, as well as lowered
N-acetylaspartate signal-to-noise ratios in cortical and subcortical regions, which was interpreted as possible neuronal atrophy and degeneration. The use of signal-to-noise ratios and limited spatial resolution were notable limitations of this study, as more recent MRS studies have analyzed the ratios of metabolites
(85). Potts et al.
(86) showed in another MRS study that patients with social anxiety disorder had a greater decrease in putaminal volumes during aging than normal comparison subjects. In studies of cerebral blood flow (CBF), Stein and Leslie
(87) found no basal metabolic cerebral differences between patients and comparison subjects on SPECT, which indicated that any posited subcortical abnormality might not affect resting metabolism. Bell et al.
(88), in a symptom-provocation study measured by means of H
215O-labeled PET, reported an array of anxiety-related changes but stated that the changes specific to social anxiety disorder included increased regional CSF in the right dorsolateral prefrontal cortex and left parietal cortex. Finally, a recent fMRI study
(89) implicated the amygdala in the pathophysiology of social anxiety, suggesting the generation of a hypersensitive amygdala when patients are exposed to potentially fear-relevant stimuli. In this study, neutral face stimuli elicited greater amygdala activity bilaterally in patients versus comparison subjects, despite knowledge that the neutral faces were not harmful, as shown by subjective ratings of anxiety. The causal relationship between fear elicitation and amygdaloid activation is unclear; however, this preliminary study is the first direct evidence for a role of the amygdala in social anxiety disorder.
In summary, there are few replicated neuroimaging studies to date regarding social anxiety disorder, but the convergence of data thus far implicates basal ganglia structures, the amygdala, and varied cortical regions. SPECT studies of the dopamine transporter and D
2 receptor in the striatum thus far are inconclusive in confirming a hypothesis of low dopamine innervation. Recent initiatives, such as the development of a PET D
2 receptor agonist ligand
(90), which allows for direct determinations of neurotransmitter-D
2 receptor interactions, will potentially provide valuable information on the role of this receptor in social anxiety disorder.
Conclusions
There are many unanswered questions regarding the neurobiology of social anxiety disorder. Given our assertion that social anxiety disorder should be conceptualized as a chronic neurodevelopmental illness beginning in childhood, several issues require further inquiry. First, we have no knowledge of studies examining the use of early identification and treatment of social anxiety disorder and its comorbid disorders and childhood precursors. Childhood social anxiety disorder is often comorbid with generalized anxiety disorder or separation anxiety disorder
(91), and these comorbid forms of the illness have a greater association with panic disorder
(92). Comparisons of laboratory neurobiological and neuroimaging measures of successfully treated patients with early intervention and successfully treated patients who were managed only in adulthood would be of interest, as would analyses of treatment responsivity across comorbid subgroups. Such secondary prevention studies might be the natural extension of longitudinal studies of behaviorally inhibited children.
Second, a better understanding of the developmental neurobiology of the brain regions important in social anxiety, such as the amygdala and striatum, and their interactions with the cortex, ascending monoaminergic systems, and hippocampus, clearly is necessary. Related to this objective neurodevelopmental genetic research, we should attempt to target susceptibility genes for the broad social anxiety phenotype. We have a limited understanding of the interaction between genetic vulnerability and stress exposure in socially anxious individuals. Cross-fostering paradigms in which primates raised under the variable-foraging-demand condition are randomly assigned to the offspring of either socially withdrawn or socially competent mothers might help answer the question of whether stress exposure has a more pernicious effect on genetically susceptible individuals.
Third, MRS imaging can be used to study neurotransmitter systems that have not received extensive attention in social anxiety, such as the glutamatergic system. Preclinical rodent models contend that prefrontal cortical efferents, either directly or by means of thalamic nuclei efferents, use the glutamatergic system as a primary source of neuronal stimulation of the “fear” neurocircuitry, which originates from the central nucleus of the amygdala and bed nucleus of the stria terminalis
(93,
94). Stressful situations faced by a person with social anxiety disorder might stimulate glutamate release in hippocampal
(38) and other brain regions. In this light, agents that attenuate glutamatergic neurotransmission should reduce anxiety levels, as well as the concomitant biochemical alterations associated with stress. Clinical investigations of glutamatergic antagonists might be warranted, since the SSRIs have been only partially successful in the treatment of this disorder. MRS also allows investigators to explore neurotransmitter interactions in vivo, such as the interaction between serotonin and glutamate, elegantly recently explored by Rosenberg et al.
(95) in pediatric OCD.
Finally, an important limitation of our understanding of the neurobiology of social anxiety is the difficulty in discriminating what findings are a response to anxiety or stress and what are true risk factors for the development of anxiety. It is of importance that the clinical neuroendocrinology of social anxiety suggests a fully compensated state in adulthood, in that no peripheral (i.e., HPA axis) pathology is evident. In this light it would be of interest to study patients with a recent onset of social anxiety disorder versus patients with distant onset in order to gauge which neuroendocrine findings persist and which ones change over the course of the illness. Another important contrast would be to study patients with active social anxiety disorder versus patients in remission. A more refined understanding of this compensatory phenomenon might offer valuable insights not only into social anxiety disorder but into other psychiatric disorders with prominent neuroendocrine abnormalities as well.