Not until the introduction of clozapine has there been any consistent documentation indicating superiority of any one antipsychotic for the treatment of schizophrenia
(1,
2). Unlike the traditional antipsychotics, which have been postulated to exert their pharmacological effects primarily by means of D
2 receptor antagonism, clozapine’s clinical efficacy is thought to be, in part, the result of serotonin 2A receptor antagonism. This difference in pharmacodynamics, combined with its low propensity to cause extrapyramidal symptoms or elevate prolactin, has resulted in clozapine’s categorization as an “atypical” antipsychotic, a designation that continues to be more of a working concept than a well-defined and validated classification. Although the atypical characteristics of clozapine were enthusiastically welcomed, the clinical use of clozapine was initially curtailed after reports of agranulocytosis in the 1970s
(3–
5). The use of clozapine has been further hindered by its associated adverse effects, which include weight gain
(6,
7), seizures
(8), and serum lipid abnormalities
(9–
13).
Investigators have shown that, despite no observed changes in total cholesterol, clozapine-treated patients have significantly higher follow-up serum triglyceride concentrations than they had before taking the drug
(9–
13). Apart from the obvious health risks associated with dyslipidemia, there is the potential that alterations in serum lipids may have a substantial impact on clozapine’s plasma distribution and subsequent clinical effectiveness. In this regard, studies have demonstrated that interactions with plasma lipoproteins can modify the pharmacokinetics, tissue distribution, and pharmacological activity of lipophilic compounds
(14–
18). The fact that clozapine is a hydrophobic compound makes it intriguing, if not essential, to examine its association with plasma lipoproteins. Therefore, the objective of this study was to determine if dyslipidemia results in a change in clozapine’s plasma distribution.
Method
To gain an understanding of how lipids influence the plasma distribution of clozapine, [
3H]clozapine plus cold clozapine (335 ng/ml) were incubated in plasma samples with varying total cholesterol, lipoprotein cholesterol, and triglyceride concentrations for 10 minutes at 37°C. Following incubation, the plasma was cooled to 4°C and separated into its lipoprotein and lipoprotein-deficient fractions by density gradient ultracentrifugation as described elsewhere
(19). Very-low-density lipoprotein, low-density lipoprotein, high-density lipoprotein, and lipoprotein-deficient (which consists of primarily albumin and α
1-acid glycoprotein) fractions were analyzed for [
3H]clozapine against external standard calibration curves (corrected for quenching and luminescence). Total cholesterol, lipoprotein cholesterol, triglyceride, and protein concentrations were determined with enzymatic assay kits as described elsewhere
(19).
The plasma distribution of clozapine was replicated four times with each of the six plasma samples. Correlations between clozapine recovery and total cholesterol or triglyceride concentrations were analyzed by using the Pearson correlation coefficient test. A one-way analysis of variance was performed to examine clozapine recovery in the different plasma fractions. The percentage of clozapine recovered in a specific fraction (e.g., very-low-density lipoprotein) was compared between the six different plasma samples (i.e., six levels for each comparison). Critical differences were identified by using Tukey-Kramer post hoc tests and were considered significant if p<0.05. All data are expressed as means and standard deviations.
Results
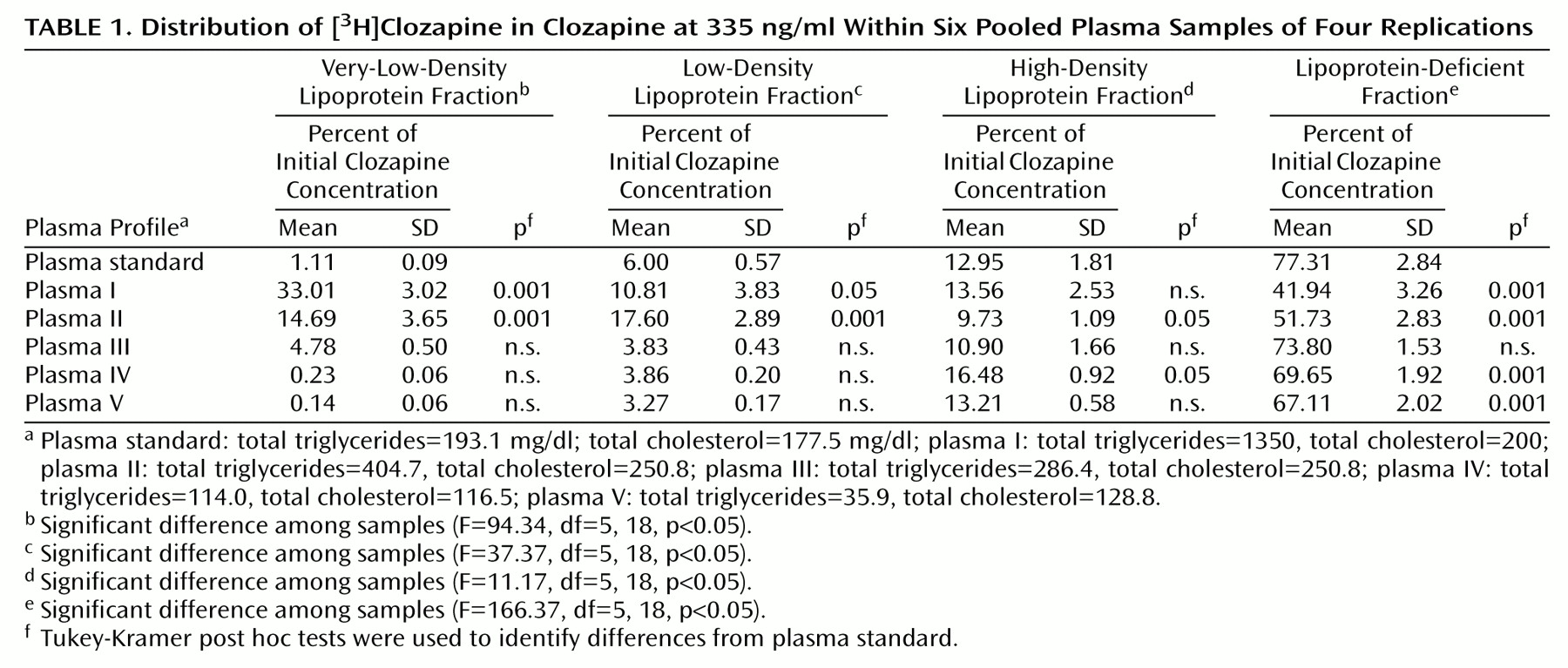
Compared with the plasma standard, significantly more clozapine was recovered in the very-low-density lipoprotein fraction of plasma samples with elevated total cholesterol and triglycerides (plasma I and plasma II) (
Table 1). Correlation analysis revealed a positive correlation between total plasma triglyceride concentration and clozapine recovery from the very-low-density lipoprotein fractions (r=0.98, df=22, p<0.05). Similarly, clozapine recovery was noted to be higher in the low-density lipoprotein fractions of plasma samples I and II (
Table 1). However, unlike the very-low-density lipoprotein fraction, clozapine recovery in the low-density lipoprotein fraction did not correlate with either total plasma cholesterol or triglyceride concentration. This redistribution of clozapine into the very-low-density lipoprotein and low-density lipoprotein fractions coincided with significantly lower values for clozapine recovery in the lipoprotein-deficient fraction of plasma samples containing higher total plasma cholesterol and triglyceride levels (plasma I and plasma II). Finally, 3%–6% of unbound clozapine was collected in the lipoprotein-deficient fractions (data not shown), which is consistent with the product monograph for clozapine
(20).
Discussion
The major finding of this study is that clozapine redistributes itself from the lipoprotein-deficient fraction to the very-low-density lipoprotein fraction as plasma triglyceride levels increase. Given that this redistribution will likely influence the kinetics of clozapine, it provokes the question as to what effect, if any, this phenomenon has on clinical outcome. First, one needs to take into consideration whether this redistribution affects the free plasma concentration of clozapine. Examination of the lipoprotein-deficient fractions (data not shown) does not indicate any reduction in the unbound, free plasma concentration of clozapine. Furthermore, it is possible that clozapine’s association with the very-low-density lipoprotein and/or low-density lipoprotein fractions may involve solubilization rather than binding. If this is the case, then one could expect an increase in the free plasma concentration of clozapine.
Apart from the potential influence on free plasma concentrations of clozapine, one must also consider what effect the redistribution of clozapine has on its ability to penetrate the blood-brain barrier. Made of lipophilic endothelial cells, the blood-brain barrier moderates the passage of compounds from the blood into the brain’s interstitial tissues. The rate of transport of compounds across this physiological barrier is dependent on molecular size, density, and the lipophilic nature of the agent
(21). It is conceivable that clozapine’s association with the very-low-density lipoprotein fraction of human plasma leads to an increased receptivity toward the blood-brain barrier. This can be rationalized by the fact that clozapine’s interaction with the very-low-density lipoprotein fraction would increase its lipophilicity, a property that aids in transport across the blood-brain barrier.
On the basis of these theoretical arguments, we hypothesize that the elevated triglycerides observed in clozapine-treated patients
(9–
13) may result in an increased passage of clozapine across the blood-brain barrier. This would result in more clozapine reaching the site of action and possibly increasing its pharmacological effectiveness. Indirect support for this hypothesis comes from two studies. In the first study
(22), investigators noted significant improvements in BPRS total score and composite negative symptoms score for patients with marked clozapine-induced weight gain. Although the investigators in this study did not examine lipoprotein profiles, it is reasonable to believe that the weight gain observed coincided with elevated triglyceride levels as previously reported
(9–
13). In a smaller, prospective study
(10), clozapine-treated patients demonstrated significant improvement in BPRS scores as well as significant increases in triglyceride levels. Although these preliminary data are limited by the small number of plasma samples, they do expose the concept that dyslipidemia may affect pharmacological activity. Apart from the effect of dyslipidemia on pharmacological activity, the clinical significance of this phenomenon also requires further study.