Several neurochemical hypotheses have been proposed to explain the origin of schizophrenia, including abnormal dopamine, serotonin (5-HT), γ-aminobutyric acid (GABA), and/or glutamate neurotransmission in different regions of the brain
(1–
8). Abnormalities in the dorsolateral prefrontal cortex have figured prominently in many of these hypotheses, in part due to results of in vivo imaging and neuroanatomical studies
(9–
11), although there is evidence for structural, metabolic, and neurochemical abnormalities in many other brain regions, including the thalamus, the hippocampus, and the cingulate and entorhinal cortices
(1,
12–
19).
Strong evidence supporting an association between glutamatergic hypofunction and schizophrenia has come from pharmacological studies showing that
N-methyl-
d-aspartic acid (NMDA) receptor antagonists such as phencyclidine and ketamine can induce many of the psychotic signs and symptoms of schizophrenia in normal subjects, as well as exacerbate these signs and symptoms in subjects with schizophrenia
(20–
27). Observations of abnormalities in markers of glutamatergic neurotransmission in the hippocampus and the entorhinal, cingulate, orbital, and prefrontal cortices of patients with schizophrenia
(3,
13,
28–
43), the critical role of glutamatergic systems in learning and memory
(44,
45), and the clear evidence for the neurotoxicity of glutamate
(28,
46,
47) have given further credence to the potential involvement of the glutamatergic system in schizophrenia.
Glutamate receptors comprise four different receptor families: NMDA, kainate, α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA), and metabotropic receptors
(48,
49). Because glutamate is the principal excitatory neurotransmitter in the brain, its receptors are ubiquitously but relatively discretely distributed throughout the neuraxis
(50–
53). Although there is evidence suggesting that kainate receptors are predominantly presynaptic and that AMPA and NMDA receptors are predominantly postsynaptic and often coexpressed, AMPA and NMDA receptors can also be localized to presynaptic sites
(51,
54). Each receptor is assembled from multiple subunits that are encoded by different genes. The NMDA receptor is assembled from a combination of four–five subunits designated NR
1 and NR
2A–D. The NR
1 subunit is often considered obligatory for functional NMDA receptor assemblies, but the remaining subunits can vary. Additional heterogeneity is conferred by eight alternative splice variants of the NR
1 subunit that have different expression patterns within the human brain
(55). The pattern of subunits assembled to form specific glutamate receptors varies from brain region to brain region
(55). In the cerebral cortex and the hippocampus, NMDA receptors are assembled from NR
1, NR
2A, and NR
2B subunits, and the NR
2C and NR
2D subunits are generally thought not to be expressed at appreciable levels
(54,
56) (but see reference
38). Since the various combinations of NMDA receptor subunits confer different sensitivities to various endogenous and exogenous glutamatergic ligands and since NMDA receptors can be localized pre- and postsynaptically
(57–
59), the release of glutamate from any given neuron or a pharmacologic glutamate ligand can have a wide range of effects in different brain regions and on glutamatergic function and activity.
At a postsynaptic level, the clustering, assemblage, and anchoring of NMDA receptors is governed in part by a protein family known as postsynaptic density 95/synapse-associated protein 90 (PSD-95/SAP-90) that appears also to play an important role in the binding and assemblage of signal transduction complexes
(60–
65). PSD-95 is exclusively localized with postsynaptic NMDA receptor-related densities
(62,
66), with significantly greater association with the NR
2A and NR
2B subunits than with the NR
1 subunit, although it does co-localize and associate with some splice variants of the NR
1 subunit
(60,
62). PSD-95 also plays an important role in signal transduction, nitric oxide neurotoxicity linked to NMDA receptor activation
(67,
68), and NMDA-induced long-term potentiation
(69). Because of these characteristics, PSD-95 is important in providing a functional scaffold for postsynaptic NMDA receptors and in mediating intracellular NMDA receptor functions.
The study described here sought to determine whether the expression of genes encoding for NR
1, NR
2A, and NR
2B subunits of the NMDA receptor and their postsynaptic anchoring protein PSD-95 are specifically altered in the dorsolateral prefrontal cortex of patients with schizophrenia. The dorsolateral prefrontal cortex was studied in postmortem brains of elderly schizophrenic subjects who had been antemortem diagnosed with DSM-IV criteria
(70–
72), had no other neuropsychiatric disease, had died of natural nonviolent causes without coma, had no evidence of significant neuropathology
(73), and had clear documentation of neuroleptic drug exposure during the months and weeks before death. Specimens of the dorsolateral prefrontal cortex from these subjects were compared to identically treated and dissected specimens from normal elderly subjects who were found by chart review and caregiver interviews to have no neuropsychiatric or neurological diseases and who had no significant neuropathology. Specificity to schizophrenia was tested by the inclusion of specimens from patients with Alzheimer’s disease. To determine the specificity of findings to the dorsolateral prefrontal cortex, specimens from the primary occipital cortex (Brodmann’s area 17) from the same subjects were dissected and assessed for NMDA receptor and PSD-95 transcript mRNA abundance by using identical procedures.
Results
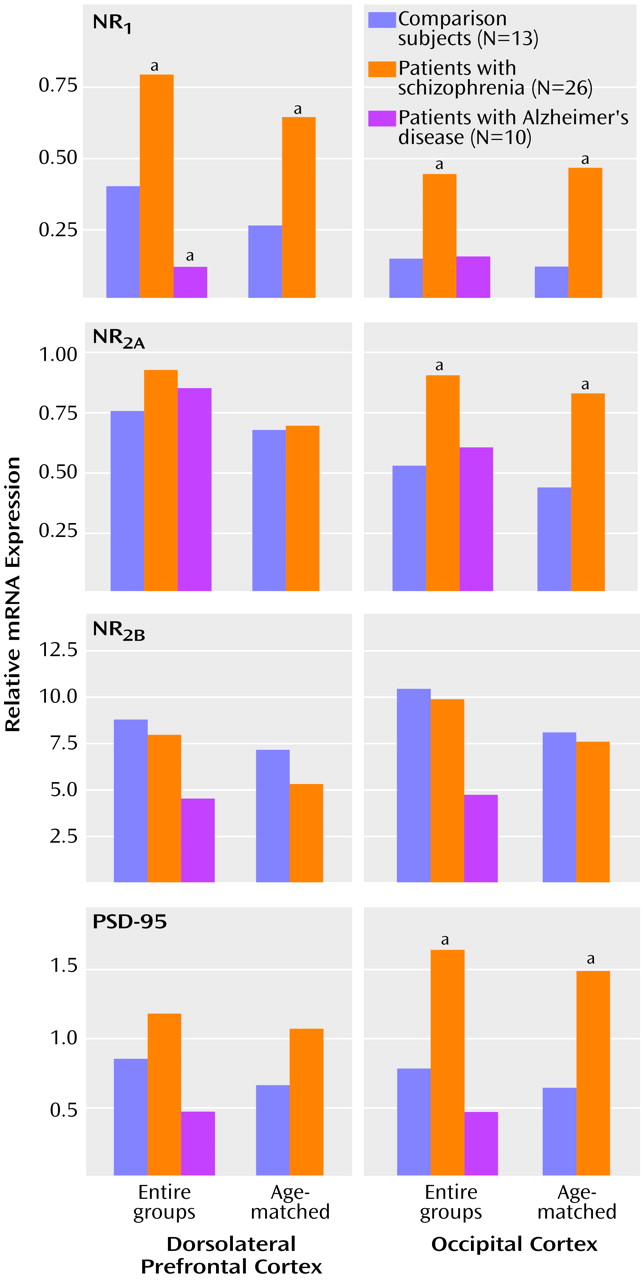
The relative abundance of NMDA receptor subunit expression in the dorsolateral prefrontal cortex and the occipital cortex for each group is shown in
Figure 2. NR
1 subunit expression in the dorsolateral prefrontal cortex was significantly higher in the schizophrenia group and significantly lower in the Alzheimer’s disease group, compared with the normal elderly group (F=12.75, df=2, 46, p=0.00004) (Newman-Keuls tests: schizophrenia versus normal elderly, p=0.009; Alzheimer’s disease versus normal elderly, p=0.05; Alzheimer’s disease versus schizophrenia, p=0.0002). Higher levels of NR
1 subunit expression were evident in the occipital cortex of the schizophrenia subjects than in the normal subjects, but not in the Alzheimer’s disease subjects (F=6.79, df=2, 46, p=0.003) (Newman-Keuls tests: schizophrenia versus normal elderly, p=0.02; Alzheimer’s disease versus normal elderly, p=0.94). Expression of NR
2A in the dorsolateral prefrontal cortex was not significantly different in either the schizophrenic or Alzheimer’s disease subjects relative to normal elderly subjects (p>0.14, Newman-Keuls). However, the level of NR
2A expression in the occipital cortex of the schizophrenic subjects was significantly higher than that of the normal elderly subjects (F=5.5, df=2, 46, p=0.007; p=0.04, Newman-Keuls). The lower level of NR
2A expression in the occipital cortex of the Alzheimer’s disease group, relative to the normal comparison group, did not reach statistical significance (p=0.40, Newman-Keuls). The expression of the NR
2B subunit was not significantly altered in any group in either of the brain regions examined (F<0.9, df=2, 46, p>0.4). The expression of the mRNA for PSD-95 was also significantly higher in the schizophrenia subjects than in the normal elderly subjects (
Figure 2). The schizophrenia-related difference in the expression of PSD-95 was most evident in the occipital cortex (F=4.31, df=2, 46, p=0.02; schizophrenia versus normal elderly, p=0.04, Newman-Keuls) but did not reach statistical significance in the dorsolateral prefrontal cortex (p=0.31, Newman-Keuls). PSD-95 gene expression was unchanged in the brains of the Alzheimer’s disease cohort.
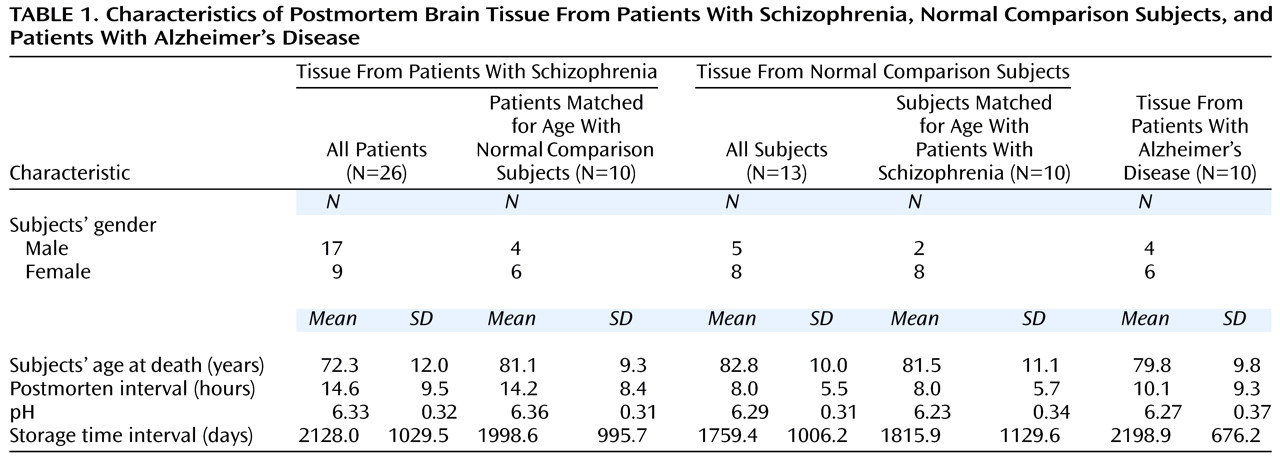
The schizophrenic cohort was significantly younger than the normal elderly group (
Table 1), and age at death correlated significantly with the expression of the NR
1 and NR
2A subunits of the NMDA receptor (r<−0.31, df=47, p<0.04) when the entire cohort was considered. The expression of the NMDA receptor subunits and PSD-95 mRNA did not correlate significantly with age within the schizophrenia cohort despite a relatively broad age range of 52–97 years. Two approaches were taken to determine whether age at death affected the differences between groups. First, ANCOVAs, with age as a covariate, were done. The inclusion of age as a covariate did not alter the pattern or the statistical significance of the results in any way (e.g., the ANCOVA comparing NR
1 expression in the dorsolateral prefrontal cortex was significant [F=9.6, df=2, 45, p=0.0003]). Second, the schizophrenic and normal elderly groups were subgrouped into two groups of 10 subjects each, matched for age to within 1 year. The differences in the expression of the NMDA receptor subunits and PSD-95 were reassessed by using t tests. As for the ANCOVA, the same significant group differences (t<−2.2, df=18, p<0.04) found when analyzing the entire cohort were observed when comparing the age-matched schizophrenic and normal elderly subjects.
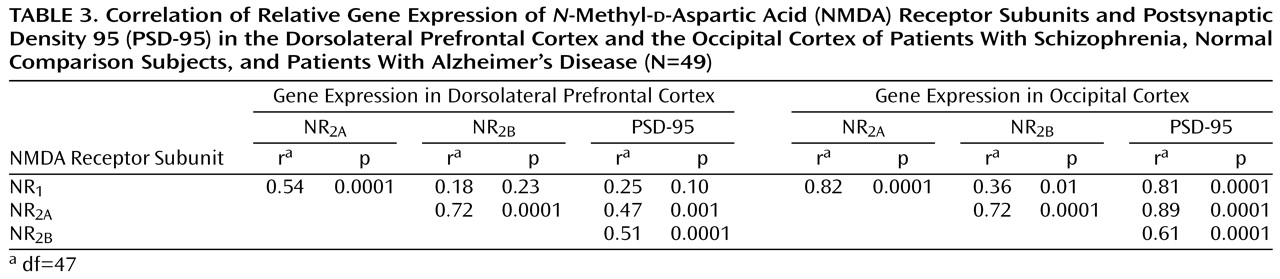
Table 3 shows the correlation of expression of the different NMDA receptor subunit mRNAs with each other and with PSD-95 in each brain region for the entire cohort. Nearly identical results were obtained when separate correlational analysis was performed for the schizophrenia subjects, the Alzheimer’s disease subjects, and the normal elderly subjects. Similarly, these correlations were not unduly influenced by differences in the age of the subjects, since the results were nearly identical when the contribution of age was factored out by using partial correlation analyses. PSD-95 gene expression correlated best with the expression of NR
2A and NR
2B in the dorsolateral prefrontal cortex and with NR
1 gene expression in the occipital cortex but not with NR
1 gene expression in the dorsolateral prefrontal cortex. Comparison of the correlations of PSD-95 with NR
1, NR
2A, and NR
2B expression in the dorsolateral prefrontal cortex versus the occipital cortex (using Fisher’s r-to-z transformation
[86]) showed that the correlations of PSD-95 with NR
1 and NR
2A were significantly stronger in the occipital cortex than in the dorsolateral prefrontal cortex (NR
1: t=5.5, df=47, p<0.001; NR
2A: t=4.1, df=47, p<0.001).
All of the schizophrenic subjects had been exposed to neuroleptics for decades. As mentioned previously, the history of neuroleptic exposure for each subject was assessed in detail by examining his or her medical chart. Of the 26 schizophrenic subjects, 13 had been exposed to neuroleptics to within 1 week of death, while neuroleptic medications had been discontinued for the remaining 13 subjects from 1 week before death to as long as 124 weeks before death. The neuroleptic-free interval did not correlate with the expression of any of the genes studied (r=–0.16 to 0.11, df=24, p>0.42). To further assess the possible influence of acute neuroleptic exposure on NMDA and PSD-95 gene expression, the schizophrenic group was subdivided into those who had been exposed to neuroleptics to within 6 weeks of death (N=16) and those who had been neuroleptic free more than 6 weeks (N=9) (data missing for one subject). Comparison of NR1, NR2A, NR2B, and PSD-95 gene expression in the dorsolateral prefrontal cortex and occipital cortex of these two groups did not reveal any significant differences (t tests, df=23, p>0.19, data not shown). As a further test of the potential influence of neuroleptic exposure on the expression of the genes of interest, NR1, NR2A, and NR2B gene expression was compared in the cortices of rats that had been treated with a daily 2-mg/kg dose of haloperidol for 3 weeks. No significant differences in NR1, NR2A, and NR2B gene expression were detected in the cortices of rats treated with haloperidol versus saline-treated rats (t<1.4, df=14, p>0.19).
Discussion
The results of this series of studies have shown that the expression of genes encoding for the predominant cortically expressed subunits of the NMDA receptor is abnormal in the dorsolateral prefrontal cortex and occipital cortex of schizophrenic subjects hospitalized for the long term who died of natural causes in old age. The expression of the NR
1 subunit was consistently higher in both regions of the cortex in the schizophrenic subjects than in normal elderly comparison subjects, and the expression of the NR
2A subunit was nominally higher in the dorsolateral prefrontal cortex and significantly higher in the occipital cortex in the schizophrenic subjects. The expression of the NR
2B subunit was not significantly altered in either region in the schizophrenic subjects. In the schizophrenic subjects, the higher level of NMDA receptor subunit gene expression was accompanied by comparable higher levels of cortical-region-specific expression of PSD-95, which is associated with postsynaptic NDMA receptor specializations. This dysregulation in NMDA receptor subunit expression was specific to the schizophrenic subjects, insofar as identically treated and studied specimens from Alzheimer’s disease patients showed lower than normal levels of NR
1 expression, as expected from previous studies
(87,
88). Replication of the finding of lower levels of NR
1 expression in Alzheimer’s disease confirms the validity of the methods used in this study and adds to the reliability of the findings in the schizophrenic tissues.
The altered expression pattern of the NMDA receptor subunits is unlikely to have been a result of the younger age of the schizophrenia group, because group differences persisted in analyses that included age as a covariate and were evident even when subgroups of schizophrenics and normal elderly subjects were matched closely for age. In addition, age did not correlate with the expression pattern of any of the genes in the schizophrenia cohort, despite those subjects’ relatively broad age range. It is possible that the observed differences between the normal elderly subjects and schizophrenic subjects resulted not only from schizophrenia but from an interaction between schizophrenia and the age of the subjects. Only replication of the study in a younger cohort can address this question. Similarly, it is noteworthy that the schizophrenic subjects in this study were chronically and severely ill and had required hospitalization for most of their lives. Whether the observed changes in NMDA receptor subunit and PSD-95 gene expression will generalize to less severely affected subjects is another question that must await replication of the study in a less severely affected cohort.
The possibility that the observed differences in NMDA and PSD-95 gene expression were influenced by exposure to neuroleptics cannot be excluded, but it is unlikely that the higher levels of NMDA receptor subunit gene expression were due to acute neuroleptic effects. Gene expression did not correlate with the amount of time subjects had been medication free before death; it was not differentially affected when the schizophrenia group was stratified into subgroups who had taken neuroleptics until the time of death versus those who had been neuroleptic free for 6–124 weeks; and it was not observed in the cortices of rats treated subchorionically with haloperidol for 3 weeks. That the observed NMDA receptor subunit gene expression was unlikely to have been directly influenced by neuroleptic exposure is supported further by other studies that have failed to find increases in cortical NMDA receptor gene expression after neuroleptic treatment
(89–
91).
The complexity of the glutamate system and its receptors is paralleled by the complexity of findings with respect to the expression of glutamate receptors in the brain in schizophrenia. Different studies have reported different findings in different regions of the brains of schizophrenic subjects. One study, which used in situ hybridization techniques and specimens from the same collection used here, reported higher levels of NR
1 and NR
2A expression in the prefrontal cortex of schizophrenic subjects
(37), substantiating the results reported here with different detection techniques and different molecular probes. Other studies have reported higher levels of glutamate receptor binding in the orbital frontal cortex and the superior temporal gyrus that are in general agreement with the current findings
(34,
35,
41). Results from another in situ hybridization study found that while the overall abundance of NMDA receptor subunits did not differ in the frontal cortices of schizophrenic subjects relative to comparison subjects, there was a shift toward increased abundance of the NR
2 subunit class, especially NR
2D subunits
(38). Recently, Gao et al.
(42) reported lower NR
1 expression and higher NR
2B expression in several subregions of the hippocampus of schizophrenic subjects, whereas the expression of the NR
2A subunit was unchanged in the same regions. These and other studies
(59) all support the conclusion that while the expression of glutamate receptors may be complex, it is nevertheless significantly affected in schizophrenia, and the expression of different subunits of NMDA receptors is significantly altered in different brain regions.
The observation of higher levels of NMDA receptor NR
1 and NR
2A subunit gene expression associated with schizophrenia raises the question of the functional consequences of this apparent change and its relationship to glutamatergic dysfunction hypotheses of schizophrenia. Knowledge of a possible disequilibrium in subunit expression does not directly provide an understanding of the attendant functional consequences, but the literature suggests that functional consequences are a likely result of altered NMDA gene expression
(59). For example, receptors assembled in vitro from the NR
1 subunit alone bind glycine antagonists, but the assembly of both NR
1 and NR
2A subunits is required for binding to glutamate antagonists and channel-blocking agents
(92). Similarly, the channel properties and antagonist affinities of NMDA receptors assembled from combinations of NR
1 and NR
2A are different from receptors assembled from the NR
1 and NR
2B subunits
(57,
58). The subunit composition of NMDA receptors can also significantly influence their susceptibility to neurotoxicity and to cell death. Cell lines transfected with NR
1/NR
2A subunits are more susceptible to cell death than those transfected with combinations of NR
1/NR
2B, which are more susceptible than cells transfected with NR
1/NR
2C subunits
(47).
The potential functional, and perhaps deleterious, consequences of higher levels of NR
1 and NR
2A gene expression in schizophrenia are further suggested by the clear evidence for increased PSD-95 gene expression and the correlation of PSD-95 gene expression with NMDA receptor subunit expression. PSD-95 was significantly overexpressed in the occipital cortex, and its expression correlated most strongly with the expression of NMDA receptor subunit mRNA in this region, although correlations between PSD-95 mRNA and NR
2A and NR
2B mRNA in the dorsolateral prefrontal cortex were also significant and relatively high. Given the importance of the C-terminal domains of NR
2A and NR
2B to receptor function and to interaction with PSD-95
(93,
94), it is likely that the NMDA receptors formed by the higher levels of NR
1 and NR
2A subunits were appropriately clustered and anchored with at least some functional integrity. The coexistence of nitric oxide immunoreactivity and NMDA receptors on cortical spiny neurons
(63,
68) and PSD-95 mediation of nitric oxide neurotoxicity induced by NMDA receptor activation
(67) raise the possibility that higher than normal levels of NMDA receptor subunit expression have detrimental consequences.
The relationship between NR1, NR2A, and NR2B subunits and PSD-95 in the dorsolateral prefrontal cortex was different than that in the occipital cortex. PSD-95 expression correlated exceptionally strongly with NR1, NR2A, and NR2B expression in the occipital cortex, but it did not correlate significantly with NR1 subunit expression in the dorsolateral prefrontal cortex despite significantly higher levels of NR1 expression in both brain regions of schizophrenic subjects. Furthermore, PSD-95 correlated less strongly with NR2A and NR2B expression in the dorsolateral prefrontal cortex than in the occipital cortex. These results suggest that NMDA receptors are expressed predominantly at postsynaptic sites in the occipital cortex but that their distribution or composition may be different in the dorsolateral prefrontal cortex. These results also raise the possibility of local regulation of glutamatergic neurotransmission and suggest that different components of the glutamatergic systems may be affected differentially in different brain regions.
The divergent correlations between the NMDA receptor subunit and PSD-95 expression in the dorsolateral prefrontal cortex and occipital cortex could imply that the pre- and postsynaptic distribution of NMDA receptors is different in the two regions. Thus, if NMDA receptors were distributed both pre- and postsynaptically in the dorsolateral prefrontal cortex, but predominantly postsynaptically in the occipital cortex, then one would expect the correlations between the NMDA receptor subunits and PSD-95 to be significantly higher in the occipital cortex than in the dorsolateral prefrontal cortex. The lack of significantly higher levels of PSD-95 expression in the dorsolateral prefrontal cortex of the schizophrenic subjects would then suggest that the observed overexpression of the NR1 subunit in that region is likely presynaptic in origin. An alteration in the balance of pre- and postsynaptic NMDA receptors in the dorsolateral prefrontal cortex of schizophrenic subjects could have broad implications, not only with respect to glutamatergic function but also relative to the responsivity to glutamatergic agonists and antagonists.
A parsimonious, yet perhaps simplistic overall interpretation of the results of this study is that some NMDA receptors are more abundant in the dorsolateral prefrontal cortex and occipital cortex of schizophrenic subjects than in those regions in comparison subjects. This interpretation is concordant with a hypoglutamatergic state hypothesis of schizophrenia, especially given the strong possibility that the increased expression of at least some of these receptor subunits is at postsynaptic sites. A traditional pharmacological interpretation would suggest that lower levels of glutamatergic activity would lead to higher levels of expression of postsynaptic glutamate receptors. In fact, animal studies have shown that NMDA receptor antagonism with phencylclidine can increase the expression of NR
1 mRNA
(95). At first glance, however, this interpretation of the results is at odds with pharmacological studies of schizophrenia with NMDA receptor antagonists. Studies of the psychotomimetic effects of uncompetitive NMDA receptor antagonists have been instrumental in the development of hypotheses that posit a hypofunctional postsynaptic glutamatergic system in schizophrenia. Some recent evidence suggests that the interpretation of the results of studies with uncompetitive NMDA antagonists may be more complex and that the symptoms induced by ketamine could be a result of increased glutamate release and/or increased activation of postsynaptic glutamate receptors
(24). Because of the presynaptic localization of some NMDA receptors
(51,
54), a hyperglutamatergic state and increased glutamate release could result from ketamine administration and its interaction with presynaptic NMDA receptor elements
(54,
96). The increased glutamate release could then act on postsynaptic glutamatergic receptors (e.g., AMPA) and provoke the psychotomimetic symptoms observed with glutamate receptor antagonists. If this interpretation of overexpressed NMDA receptors at presynaptic sites in the dorsolateral prefrontal cortex of schizophrenic patients is correct, then it would be reasonable to assume that NMDA antagonists would exacerbate the symptoms of schizophrenia. Thus, although the results of the current study and those cited earlier support the view that cortical glutamatergic systems are significantly affected and abnormal in schizophrenia, they highlight the need for further and more detailed studies to elucidate the precise nature of the glutamatergic abnormality.