Previous experiments in postmortem brain samples have demonstrated that human cocaine users have decreased striatal vesicular monoamine transporter binding sites, as assessed by the specific high-affinity radioligand dihydrotetrabenazine ([
3H]DTBZ)
(1,
2). These findings suggest the possibility that there may be damage to, or loss of, dopamine neurons in the striatum in human cocaine users. Current evidence indicates that the vesicular monoamine transporter protein (VMAT2) is a better marker of dopamine axonal integrity than the synaptic dopamine transporter, since VMAT2 exists in excessive concentrations and appears less susceptible to regulation by drug exposure
(3,
4). In contrast, dopamine transporter function is regulated in a dynamic fashion by pharmacological treatments. Cell cultures expressing cloned dopamine transporter demonstrate that treatment with cocaine or d-amphetamine regulates dopamine transporter expression in the plasma membrane
(5–
8), while postmortem studies indicate that human cocaine users display a greater number of dopamine transporter radioligand binding sites
(1,
9,
10). It has been postulated that striatal dopaminergic neurons may be injured by chronic cocaine exposure in humans, as evidenced by declines in VMAT2, dopamine transporter mRNA, and dopamine concentrations, which are accompanied by increases in dopamine transporter function intended to conserve waning supplies of neurotransmitter
(1).
Method
Subjects
Striatal specimens from cocaine-using subjects (N=35) and age-, sex-, and postmortem interval-matched comparison subjects (N=35) were assayed. The methods used to obtain brain samples have been previously described
(1) and will be reviewed briefly. Postmortem brain specimens were obtained at autopsy as authorized by the Wayne County Medical Examiner’s Office, freshly dissected, quickly frozen on dry ice, and stored at –70°C until sectioned.
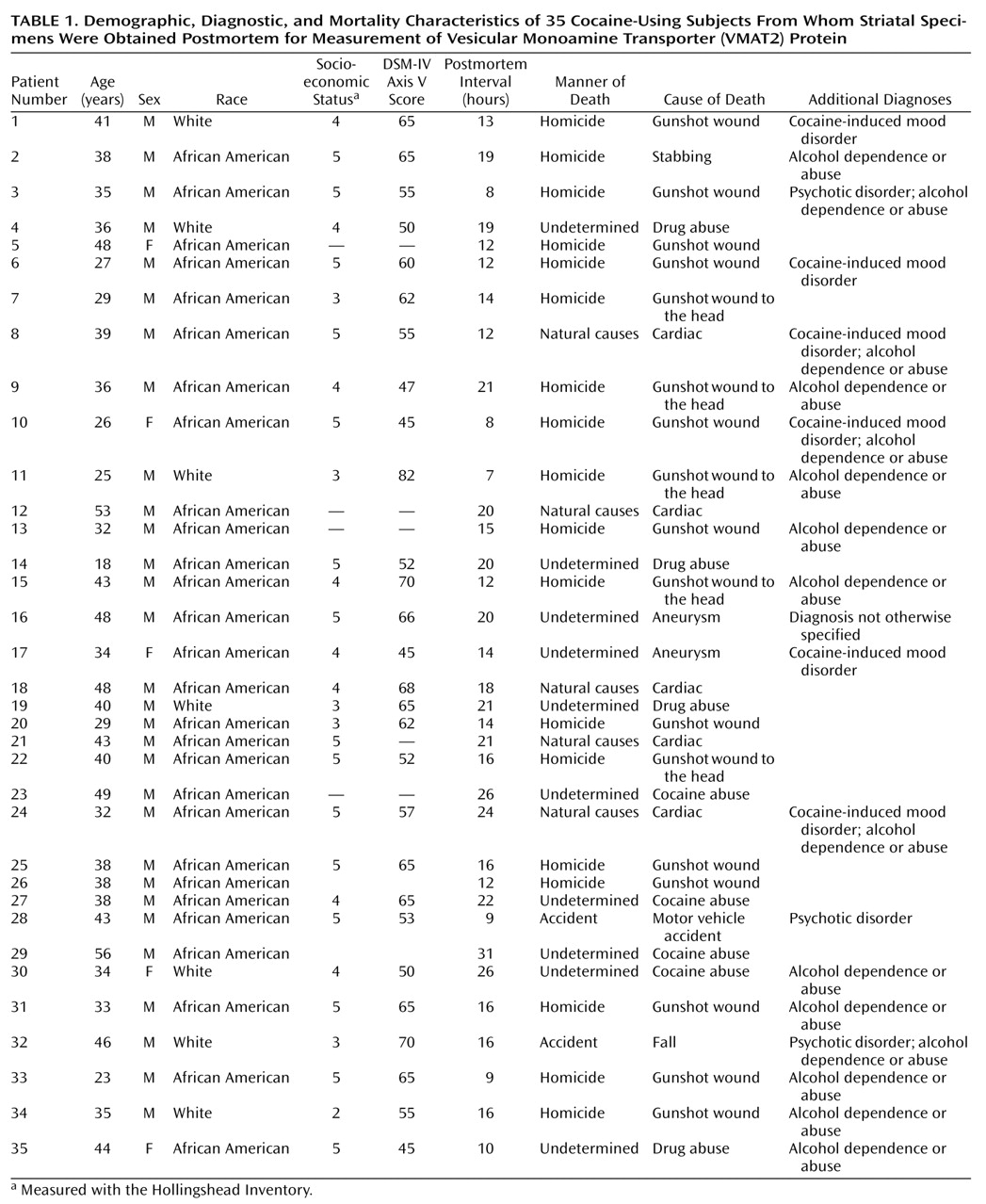
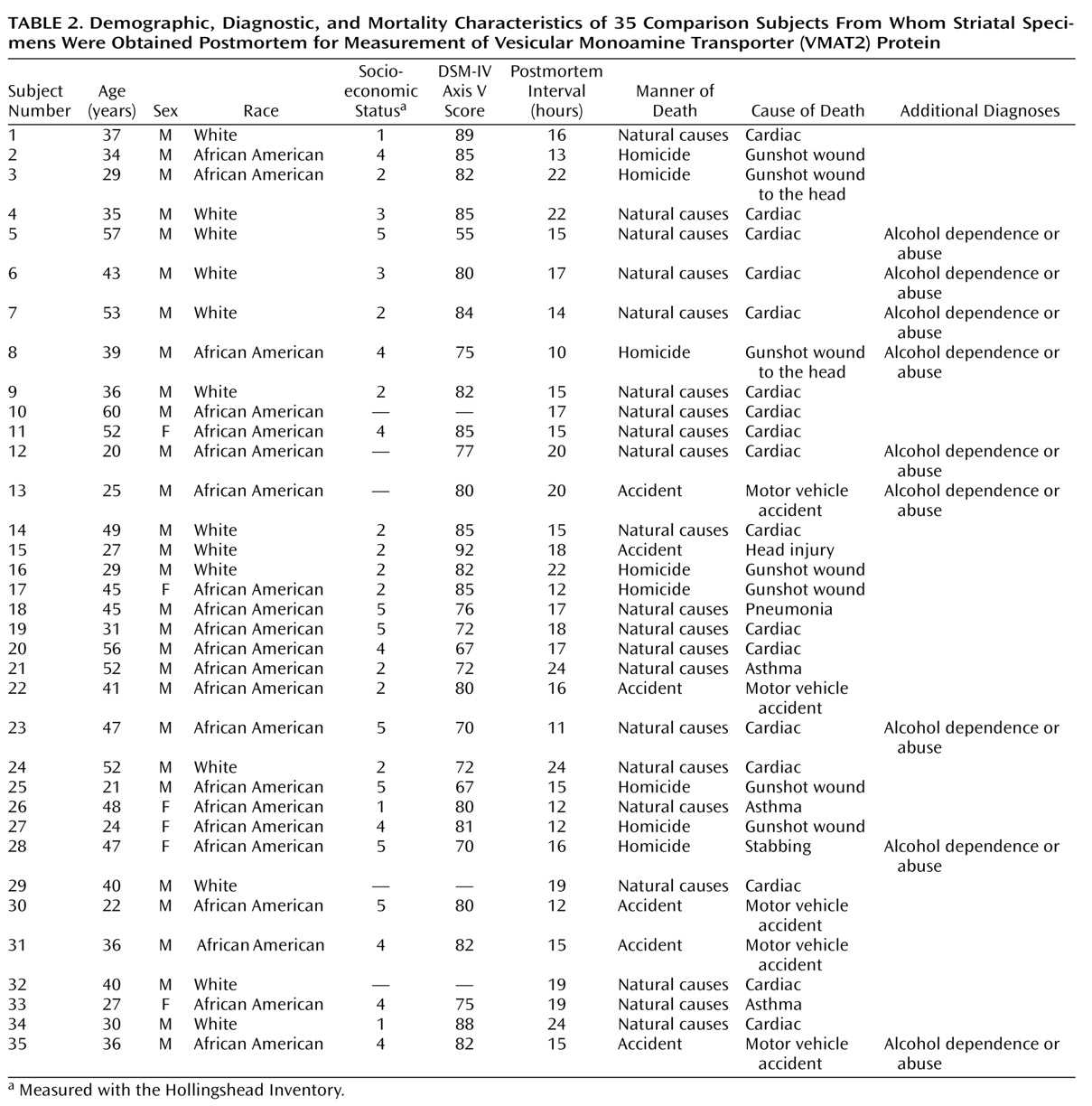
Table 1 and
Table 2 list the causes of death for each subject as well as demographic and diagnostic information. Subjects died suddenly in accidents, by assault, or of cardiovascular causes. Comparison subjects and cocaine users were matched for postmortem interval, age, sex, and race, as well as socioeconomic status to control for any nonspecific factors associated with impoverished nutrition and living conditions. The pairing of subjects was intended to result in comparable groups for group statistical analysis. Sufficient striatal samples were not available for every subject for each assay, resulting in three unpaired cocaine users being included in the VMAT2 analysis (cocaine-using subjects N=33; comparison subjects: N=30). Tissue from the right side of the brain was used for the assays. Comparison subjects met the following criteria: 1) suitable method of death (rapid, not result of chronic condition); 2) appropriate age, sex, and race to balance the cocaine-using subjects; and 3) absence of DSM-IV diagnoses, other than ethanol use or nicotine use. Cocaine users were without evidence of chronic medical illness and exhibited a range of typical psychiatric symptoms and psychoactive substance use disorders.
Clinical Assessment
A research psychiatrist and clinical social worker interviewed at least one knowledgeable informant for each subject, most often a first-degree relative. Other types of informants included neighbors, friends, fellow workers, police, medical examiners, physicians, mental health personnel, and newspaper reporters. Family members who participated in the study provided informed consent under guidelines approved by the Institutional Review Board of the University of Michigan. Interviews focused on ascertaining if the subject met DSM-IV criteria for any psychoactive substance abuse disorder, alcoholism, antisocial personality disorder, mood disorder, anxiety disorder, or psychotic disorder. The Hollingshead Inventory, an estimate of socioeconomic status, was derived for every subject. Axis V scores were also assigned by using the standard directions as an estimate of overall dysfunction. On the basis of available evidence, DSM-IV psychiatric diagnoses were assigned at a consensus conference. When further questions remained, the diagnostic assignment was postponed, and efforts were renewed to discover the specific information needed. Diagnoses and scores were assigned before assays were performed.
Toxicology
Urine or serum from subjects was assayed qualitatively for the presence of cocaine, opioids, antidepressants, antipsychotics, and anxiolytics by using a variety of methods, including radioimmunoassay, high-performance liquid chromatography, and gas chromatography/mass spectroscopy. Ethanol levels were measured by dichromate microdiffusion/gas chromatography methods.
VMAT2 Western Blots
Striatal samples (about 50 mg) were homogenized for 5 seconds in phosphate-buffered solution (139 mM NaCl, 2.7 mM KCl, 1.5 mM KH
2PO
4, 9.6 mM Na
2HPO
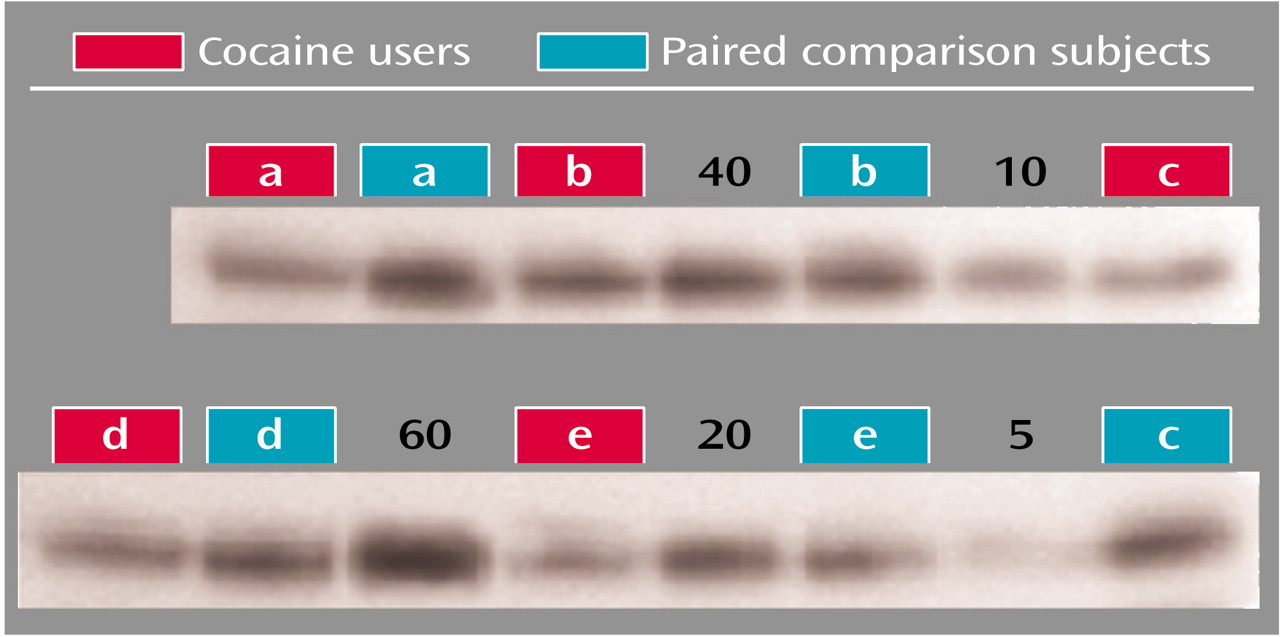
4), diluted in sample buffer, and incubated at 100°C for 5 minutes. The proteins were separated by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) with 8.75% polyacrylamide gels (8×5 cm, 2-mm thick). Each lane was loaded with 0.1 ml of sample, at a concentration of 15 mg/ml total protein determined spectroscopically by performing a DC protein assay (Bio-Rad Laboratories, Hemel Hempstead, U.K.). After separation, proteins were then electrophoretically transferred to Immobilon-P membranes at 8 V/cm for 1 hour. Blots were incubated with a blocking solution of 5% (weight/vol) dry milk in TTBS (0.1% Tween 20, 0.15 M NaCl, and 10 mM Tris-HCl [pH 7.4]) for 1 hour. After transfer, blots were probed with rabbit polyclonal antihuman VMAT2, C-terminal (1:4000 dilution; Chemicon, Inc., Temecula, Calif.) overnight followed by a horseradish peroxidase-conjugated donkey polyclonal antirabbit IgG (1:2000 dilution, Amersham Life Sciences, Piscataway, N.J.) second antibody. The antigen-antibody complex was detected by chemiluminescence (ECL Plus detection system, Amersham Life Sciences, Piscataway, N.J.). All steps were performed at room temperature. The relative optical density of protein bands was quantified by using an image analysis system (Microcomputer Imaging Devices, Ottawa, Ont., Canada). Cocaine users and paired subjects were assayed next to each other on the same gels. Two types of standards were included as controls. First, six known amounts of pooled human striatal samples, from 5 mg/ml to 60 mg/ml, were included in each gel to allow interpolation of individual values to the curve for that gel in order to account for film-to-film variations in the density of VMAT2 values obtained by interpolation to the standard curves run on each gel (
Figure 1). Second, actin levels were assessed for each individual (after stripping and reprobing each film), and total relative optical density was then adjusted as a ratio to actin for that individual. Actin levels for individual subjects were within the actin dynamic range on each gel, as all were lower compared with the density demonstrated by the 60 mg/ml known samples. Actin levels were also compared between cocaine users and comparison subjects.
High-Performance Liquid Chromatography
For the high-performance liquid chromatography assay
(15), frozen specimens were homogenized in buffer solution and then centrifuged for 15 minutes at 2°C and 17,500 g. The mobile phase buffer consisted of 0.1 M citrate, 0.075 M sodium phosphate, 0.75 mM sodium heptane sulfonate, and 17% methanol (volume for volume), adjusted to pH 4.12. A C18 chromatographic column with 5-mm microparticulate silica gel (Spherisorb ODS-2, Sigma-Aldrich Chromatography, St. Louis) was used. Separations were performed isocratically at 1.0 ml/min with a Waters (Milford, Mass.) pump and controller coupled to a model 5100A electrochemical detector (0.35 V potential) (ESA, Bedford, Mass.). Standard calibration curves were prepared by using known amounts of dopamine and plotted as peak area (microvolts per second divided by 10
–6) versus concentration of dopamine (nanograms per microliter). Retention times were distinct for each compound analyzed. Concentrations of neurotransmitters and metabolites in samples were back-calculated from the standard calibration curve and expressed as picomoles per gram of wet tissue. Chemicals, along with a grade of methyl alcohol and water appropriate for high-performance liquid chromatography, were obtained from Sigma Chemical Co. (St. Louis).
[3H]DTBZ Binding
The autoradiographic assay methods for (+)-[
3H]dihydrotetrabenazine (DTBZ) have been previously described
(1). Briefly, 16-mm-thick sections were incubated at 2°C in Tris buffer (50 mM, 120 mM NaCl, pH 7.4) for 1.5 hours. An initial saturation study indicated a dissociation constant (K
d) of 1.9 nM (SD=0.1) in human striatum. On the basis of this result, a single saturating concentration of [
3H]DTBZ (10 nM) was used to assess binding in the striatum, by means of two total and one nonspecific sections. Nonspecific binding was defined with reserpine, 10 mM. After incubation, the slides were washed and then apposed to Kodak SB-5 film for 5–7 days. Amersham tritium microscale standards (Amersham, Arlington Heights, Ill.) were co-exposed for each cassette. Optical densities were determined by using an image analysis system (Microcomputer Imaging Devices, Ottawa, Ont., Canada). Optical densities were evaluated by comparison to the Amersham standards and are expressed in nanocuries per milligram.
Data Analysis
Initially, to confirm that cocaine users and comparison subjects had been demographically well-matched, they were examined by using a one-way analysis of variance (ANOVA) that searched for age, sex, race, postmortem interval, and socioeconomic status differences. VMAT2 immunoreactivity, [3H]DTBZ binding, and dopamine levels were then compared in cocaine users and comparison subjects by univariate ANOVA, which simultaneously examined the effect of alcohol dependence as an independent variable. VMAT2 immunoreactivity, [3H]DTBZ binding, and dopamine levels were further examined for correlative relationships. Other clinical data including age, postmortem interval, sex, race, and other psychiatric diagnosis (psychosis and mood disorder) were examined for effect onVMAT2 immunoreactivity, [3H]DTBZ binding, and dopamine by using ANOVA or correlational analysis, as appropriate.
Results
Examination of subject demographics (listed in
Table 1 and
Table 2) found that there were no significant differences in age between the comparison subjects (mean=39.0 years, SD=11.2) and the cocaine users (mean=37.6 years, SD=8.6) (F=0.52, df=1, 68, p=0.57). Nor was there a difference in postmortem interval (comparison subjects: mean=16.8 hours, SD=3.8; cocaine users: mean=16.2 hours, SD=5.7) (F=0.47, df=1, 68, p=0.64). Male subjects represented 83% (N=29 of 35) of the comparison group and 86% (N=30 of 35) of the cocaine-using group (F=0.12, df=1, 65, p=0.93). African Americans comprised 63% (N=22 of 35) of the comparison subjects and 80% (N=28 of 35) of the cocaine-using group (F=2.54, df=1, 65, p=0.12). The socioeconomic status was significantly better in the comparison subjects (mean=3.3, SD=1.6) than in the cocaine users (mean=4.3, SD=0.9) (F=10.77, df=1, 57, p<0.001) as was the DSM-IV axis V score (mean=78.7 [SD=7.7] versus 59.1 [SD=9.1], respectively; t=8.997, df=58, p<0.001).
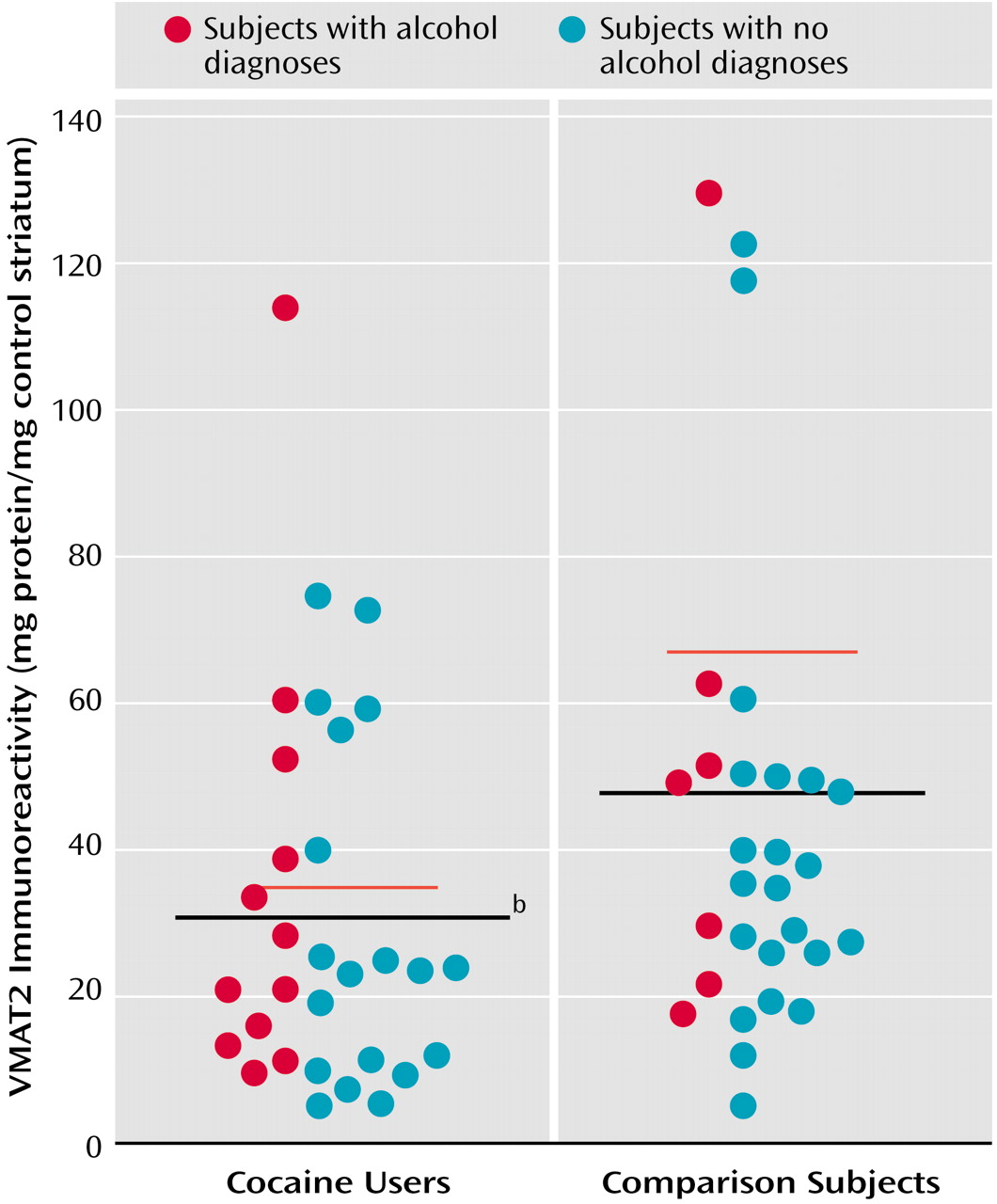
To conserve tissue for other assays, VMAT immunoreactivity was measured only in the caudate on the basis of a previous [
3H]DTBZ assay that indicated that the effects of cocaine were comparable in caudate and putamen. As seen in
Figure 2, VMAT2 immunoreactivity was significantly lower in the cocaine-using group (mean=31 mg protein/mg standard, SD=26) than in the comparison subjects (mean=48 mg protein/mg standard, SD=36). Because of the coexistence of ethanol dependence in both comparison subjects and cocaine users, the analysis also searched for possible ethanol effects. Ethanol dependence did not have an effect (F=1.58, df=1, 62, p=0.21), nor was there a cocaine-by-ethanol interaction (F=0.52, df=1, 62, p=0.47).
Nearly identical results were obtained whether the VMAT2 immunoreactivity results analyzed were those determined by interpolation to the standard curve for each gel or those determined by comparing the calculated ratios of VMAT2 relative optical densities to actin relative optical densities within individuals. An assessment of actin relative optical density levels found that they were similar in the cocaine group and the comparison subjects, indicating that cocaine exposure did not nonspecifically diminish all cellular proteins (t=0.82, df=55, p=0.42).
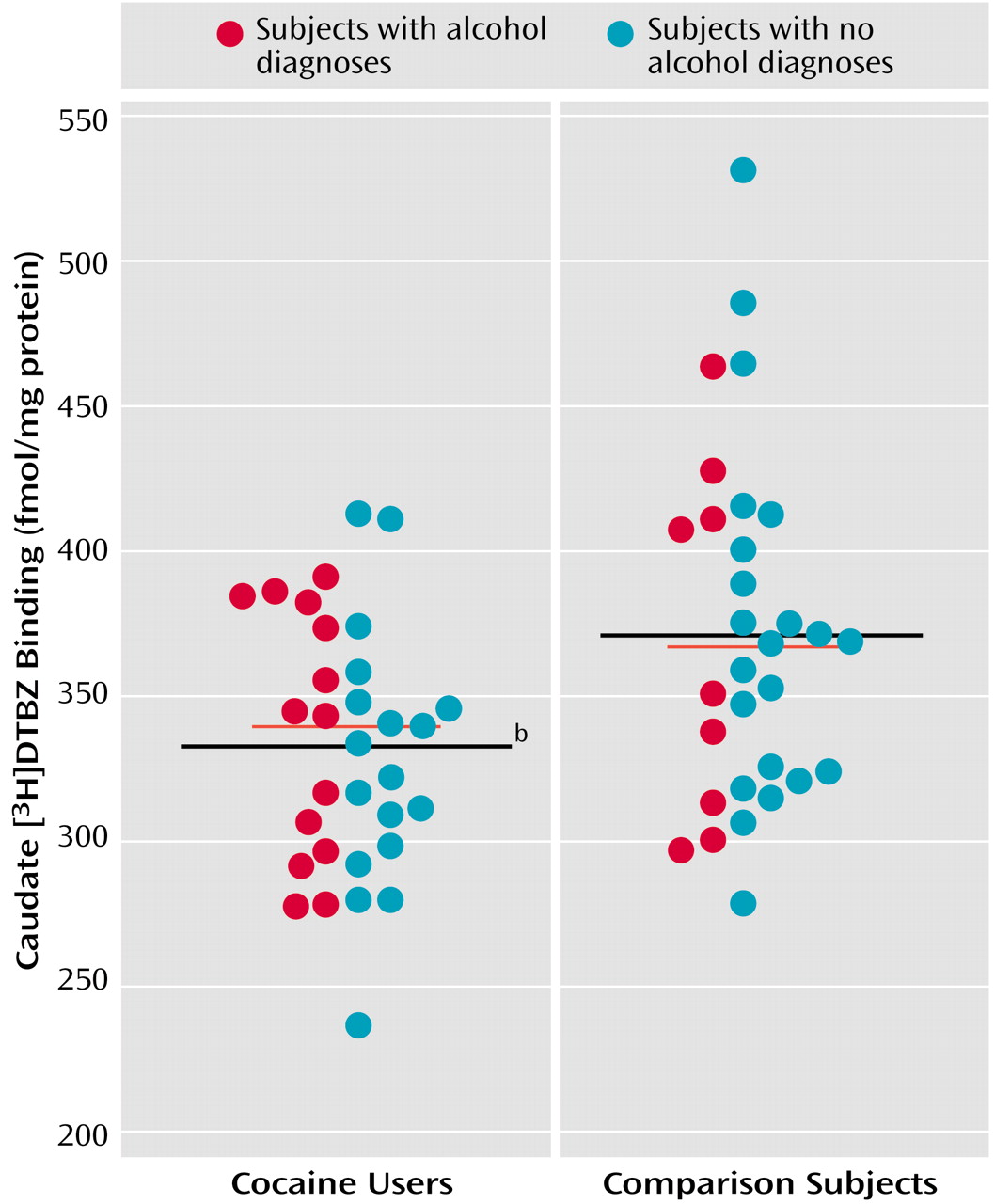
As previously detected in a subgroup of the present subjects, [
3H]DTBZ binding was significantly lower in the entire group of cocaine users in both the caudate (cocaine users: mean=334 fmol/mg protein [SD=43], comparison subjects: mean=371 fmol/mg protein [SD=60]) (
Figure 3) and the putamen (mean=336 fmol/mg protein [SD=40] and 372 fmol/mg protein [SD=49], respectively; F=10.16, df=1, 63, p=0.002). Ethanol dependence did not have an effect in the caudate (F=0.02, df=1, 63, p=0.89) or putamen (F=0.04, df=1, 63, p=0.85), nor were there cocaine-by-ethanol interactions (caudate: F=0.26, df=1, 63, p<0.62; putamen: F=1.95, df=1, 63, p<0.17).
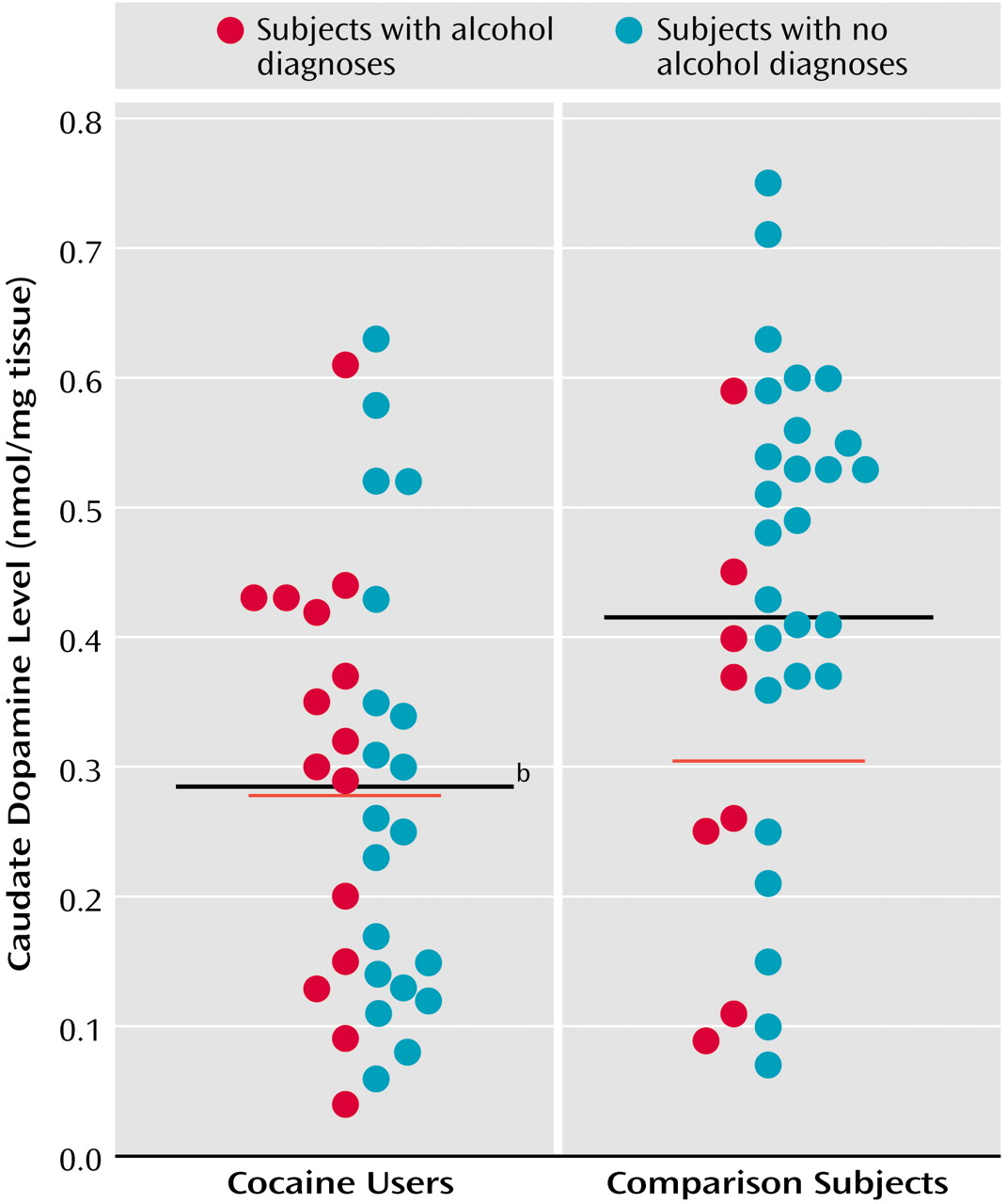
A univariate analysis found that caudate dopamine levels were significantly lower in the cocaine users (mean=0.29 nmol/mg tissue, SD=0.17) than in the comparison subjects (mean=0.42 nmol/mg tissue, SD=0.18) (
Figure 4). Ethanol dependence did not have an effect (F=1.47, df=1, 70, p=0.23), nor was there a cocaine-by-ethanol interaction (F=2.80, df=1, 70, p=0.10). The effects were not significant in the putamen, where there were fewer samples and greater variance (cocaine users: mean=0.35 nmol/mg tissue [SD=0.17], comparison subjects: mean=0.48 nmol/mg tissue [SD=0.26]; F=2.24, df=1, 59, p=0.26). In contrast to dopamine levels, HVA levels were virtually equivalent in cocaine users, alcoholics, and comparison subjects, in both the caudate and putamen.
Among the cocaine users, neither age nor postmortem interval were significantly different between those with and without an ethanol diagnosis. Likewise, neither age nor postmortem interval significantly differentiated between the comparison subjects with and those without an ethanol diagnosis. Axis V scores were nearly identical between both cocaine users with an alcohol diagnosis (mean=59.1, SD=9.2) and those without (mean=58.5, SD=11.6) (t=0.14, df=24, p=0.89). As expected, however, significantly lower axis V scores were seen in comparison subjects with an ethanol diagnosis (mean=73.4, SD=9.6) than in those without such a diagnosis (mean=80.5, SD=7.2) (t=2.13, df=26, p=0.04).
Coexisting mood and psychotic disorders were detected in a number of the cocaine users. Lower VMAT2 immunoreactivity was seen among subjects with cocaine-induced mood disorders (comparison subjects: mean=54.8 mg protein/mg standard, SD=46.0; subjects with only a cocaine diagnosis: mean=40.7 mg protein/mg standard, SD=30.9; cocaine users with additional mood disorders: mean=20.1 mg protein/mg standard, SD=17.1; cocaine users with additional psychotic disorders: mean=29.1 mg protein/mg standard, SD=5.7), but the differences among diagnostic subgroups were not significantly different (one-way ANOVA: F=2.09, df=3, 51, p=0.11). However, when the ratio of previously determined [3H]WIN 35428 binding (which is significantly greater in cocaine users) was calculated relative to VMAT2 immunoreactivity for each individual, there was a statistically significant difference among mood-disordered subjects (F=15.95, df=3, 43, p<0.0001). Tukey’s honestly significant difference post hoc tests showed that cocaine users with additional mood disorders significantly differed from comparison subjects (Q=9.77, p<0.001), subjects with only a cocaine diagnosis (Q=7.13, p<0.001), and cocaine users with additional psychotic disorders (Q=5.18, p<0.01). The overall severity of psychopathology (axis V score) in cocaine users with additional mood disorders (mean=56.1, SD=8.3) was not significantly different from that of cocaine users with no additional mood disorders (mean=59.8, SD=11.2) (t=0.87, df=24, p=0.39), suggesting that cocaine users with additional mood disorders were not more ill or dependent on cocaine. Differences in postmortem intervals and ages were statistically nonsignificant as well. [3H]DTBZ values were as follows: 338 fmol/mg protein (SD=46) in subjects with only a cocaine diagnosis, 331 fmol/mg protein (SD=39) in cocaine users with additional mood disorders, and 314 fmol/mg protein (SD=62) in cocaine users with additional psychotic disorders. There was not a significant difference in the ratio of [3H]WIN 35428 to [3H]DTBZ binding among the subgroups (F=2.95, df=3, 43, p=0.13). Dopamine values displayed no clear pattern: 0.31 nmol/mg tissue (SD=0.21) in subjects with only a cocaine diagnosis, 0.40 nmol/mg tissue (SD=0.31) in cocaine users with additional mood disorders, and 0.30 nmol/mg tissue (SD=0.11) in cocaine users with additional psychotic disorders.
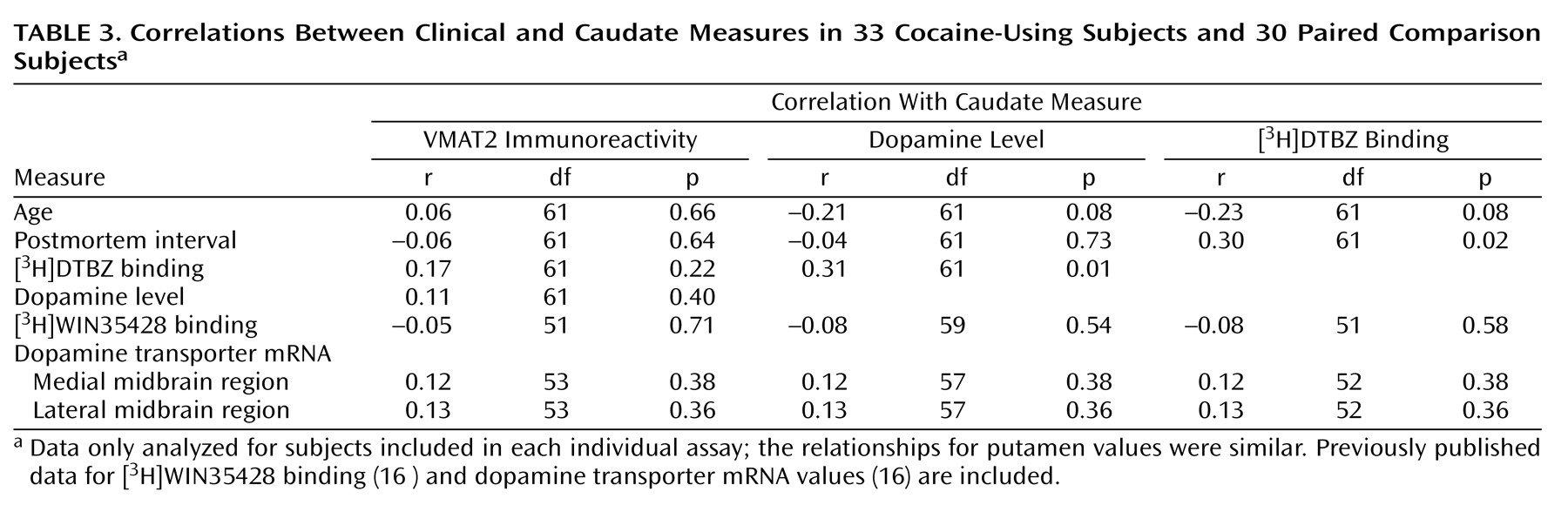
Table 3 summarizes the results from a number of correlational analyses. There was not a significant relationship between VMAT2 immunoreactivity and [
3H]DTBZ binding or between VMAT2 immunoreactivity and dopamine levels. However, dopamine level and [
3H]DTBZ binding were significantly correlated. It was found that [
3H]DTBZ binding appeared to increase with longer postmortem interval. Dopamine level and [
3H]DTBZ binding tended to decline with age, which in both cases approached statistical significance (p=0.08). VMAT2 immunoreactivity appeared less affected by age. Based on this, a review of age for various subdiagnoses found that mood disorder subjects (mean age=35 years, SD=8) and alcoholics (mean age=33 years, SD=7) were slightly younger than the remaining subjects (mean age=41 years, SD=9), indicating that the declines seen in these subjects were not caused by advanced age compared with the other subjects.
Conclusions
Striatal VMAT2 immunoreactivity and [
3H]DTBZ binding were significantly decreased in a large group of human cocaine users compared with nonusers. The present [
3H]DTBZ binding results replicate those of Wilson et al.
(2), who found a 22% decrease in [
3H]DTBZ binding in postmortem striatum from 12 severely cocaine-dependent subjects along with a concomitant loss of dopamine. The present results also complement our previous report
(16) in these same cocaine users of a mild decrease in medial midbrain dopamine transporter mRNA levels. Together, these results suggest that sustained cocaine exposure diminishes dopamine neuronal function in humans. The present results contrast to those found in a smaller postmortem study by Staley et al.
(17), who reported no [
3H]DTBZ differences in a somewhat distinct population of nine subjects specifically dying of cocaine overdose. Staley et al. also quantified antibody labeling of striatal VMAT2 in four cocaine overdoses and found a decrease that was not statistically significant relative to comparison subjects
(17).
Animal studies have consistently failed to detect evidence of neuronal loss after cocaine treatment
(3,
18–20). It is possible that the absence of neuronal effects of cocaine in previous animal experiments might reflect species differences or more intense and prolonged cocaine exposures in humans, which are difficult to replicate in animal studies
(3,
4). It is also possible that the presently described VMAT2 alterations in cocaine users might reflect compensatory adjustments in VMAT2 cellular distribution. Thus, the decline in VMAT2 could reflect a decrease in its distribution to vesicles, or even the total number of vesicles. Examination of other vesicle protein concentrations should be helpful in resolving this issue.
The lack of correlation between VMAT2 binding and immunoreactivity is puzzling. In our opinion, the complexities involved in interpreting radioligand binding data, including variable penetration to various cellular compartments, and sensitivity to modest conformational differences that are not necessarily detected as small changes in calculated affinity constants, bolster our belief in the greater validity and relative interpretive simplicity of the present protein measures. In any case, both VMAT2 measures we have performed suggest that some decline occurs overall within this population. Hogan et al.
(21) documented a similar disparity between [
3H]DTBZ binding and other measures of dopamine integrity after exposure to the toxic compound MPTP. Only when these investigators separated very light vesicles from total homogenate were they able to detect changes in [
3H]DTBZ binding in this fraction. These investigators postulate that distinct fractions of VMAT2 are plasma membrane-bound, in recycling vesicles, or are vesicles in transport and may be differently accessible compared with the mature, functional VMAT2 located in monoamine-accumulating presynaptic vesicles. Kilbourn et al.
(22) found that treatment with MPTP caused no changes in [
11C]DTBZ binding to the VMAT2 in rat striatum, despite large losses of [
3H]methylphenidate binding to the dopamine transporter. All of this leads us to infer that losses of some fraction of neuronal vesicular transporters are not always equal to other indicators of dopamine vesicular or neuronal integrity or may not occur in the same time frame. [
3H]DTBZ binding may preferentially label a subpopulation of the total VMAT2 protein detected immunologically. Thus, cocaine-induced decreases in VMAT2 could occur in neurons with overall problematic VMAT2 synthesis but relatively preserved vesicular VMAT2.
The direction of the present VMAT2 results is opposite to the effects of cocaine exposure on dopamine transporter binding sites noted in the same group of subjects. The present data suggest the possibility that loss or wastage of dopamine could induce a compensatory increase in plasma membrane uptake mechanisms. However, experiments in a model system indicate that up-regulation of the dopamine transporter can result directly from cocaine binding to the transporter, inducing upregulation independent of dopamine availability
(7,
8). Changes in dopamine transporter function may directly result from cocaine exposure, while VMAT2 regulation may reflect overall changes in dopamine metabolism.
Very low socioeconomic status scores were seen in the cocaine users, reflecting the extreme social distress many were experiencing. Although the comparison subjects were of moderately low socioeconomic status themselves, it is still possible that some unknown factors, perhaps nutritional, could be playing a role on VMAT2 beyond that caused by cocaine exposure per se. Alcohol dependence did not appear to play a role in the changes in VMAT2 and dopamine measures.
It was not possible to determine whether the presently described effects on VMAT2 measures result from short- or longer-term cocaine exposure. If short term, then differences in recent cocaine dosing and the time between the last cocaine exposure and death could account for some of the variability noted. If the effects depend on longer-term cocaine exposure, then the details of the last cocaine exposure immediately before death may be less contributory. Studies in a model system, as well as PET studies in human users, could help to determine the time course.
The statistically significant increase in [3H]DTBZ binding with longer postmortem intervals was not the usual relationship one expects. It may reflect a chance occurrence, since more than 20 correlative comparisons were performed. In any case, the cocaine user and comparison groups were well matched for postmortem intervals and postmortem interval effects did not confound the apparent difference caused by cocaine use.
Both increased [3H]WIN 35428 binding and decreased VMAT2 immunoreactivity were more likely in the same individual. One simple explanation is that these individuals had experienced greater exposure to cocaine. Alternatively, it is possible that unknown genetic factors were responsible for the increased dopamine transporter/VMAT2 ratio. Gene array studies are ongoing in samples from these same subjects to determine the pattern of genes that are altered in cocaine users.
Clinically, Schmitz and colleagues
(23) have reported that depressed cocaine users have more overall distress and poorer psychiatric functioning, as evidenced by higher scores on all subscales of the SCL-90-R, higher craving for cocaine, lower self-efficacy to refrain from drug use, and lower perceived social support. This may reflect the mediation of depressive symptoms, increasing the psychological need to use cocaine, or it could result from parallel effects on neurochemical substrates underlying both depression and craving, leading to greater use and further neuronal dysregulation. Diminished dopamine storage in conjunction with an increase in uptake by dopamine transporter could result in marked synaptic hypodopaminergia. Such an effect might explain the relationship found between increased dopamine transporter/decreased VMAT2 and depressive symptoms in the present study (albeit in a small sample of subjects).
In summary, the present results suggest that cocaine exposure in humans affects its primary neuronal target—the dopamine neuron. The present experiments could not determine if these were toxic effects, involving loss of neurons, or instead represent a complex regulatory change in VMAT2 and dopamine metabolism. In conjunction with earlier studies of the dopamine transporter in these same subjects, the present results suggest that these changes are deleterious and may contribute to depressive symptoms in some cocaine users. Recent clinical evidence indicates that cocaine use complicated by depressive symptoms carries a poorer prognosis for treatment response. Further efforts at clarifying the pathophysiological mechanisms involved may help explain treatment refractoriness and identify targets for therapeutic intervention.