Modafinil is currently approved by the U.S. Food and Drug Administration (FDA) for improving wakefulness in patients with excessive sleepiness associated with narcolepsy
(1,
2), obstructive sleep apnea or hypopnea syndrome
(3), and shift-work sleep disorder
(4) . Additional data from controlled studies suggest that modafinil may improve symptoms of fatigue and daytime somnolence in patients with Parkinson’s disease
(5) and multiple sclerosis
(6,
7) . Modafinil’s wakefulness-promoting mechanism of action is unknown. The drug appears to be distinct from psychostimulants in several respects: a selective rather than widespread brain activation
(8), a lack of addictive potential in preclinical models
(9), and an absence of clinical abuse potential
(10) .
Thus far, controlled trials of modafinil in the treatment of axis I disorders have focused on attention deficit hyperactivity disorder (ADHD), cocaine dependence, and unipolar depression. Two recent placebo-controlled studies have highlighted the efficacy of modafinil (at a mean dose of 300 mg/day) in treating children and adolescents with ADHD
(11,
12) . Data from a placebo-controlled study
(13) suggest that modafinil may decrease cocaine use in cocaine-dependent patients; in addition to manual-guided, twice-weekly cognitive behavior therapy, modafinil (at a mean dose of 400 mg/day) decreased the number of cocaine-positive urine toxicology screens and increased the rate of protracted abstinence. Although primary outcome measures have generally been negative in studies of unipolar depression in which modafinil is used for antidepressant augmentation or acceleration, secondary measures of clinical global improvement
(14) and rapid reduction in fatigue and sleepiness
(15,
16) have suggested some benefit. In a 12-week open-label extension study of adjunctive modafinil with selective serotonin reuptake inhibitors, 70% of 194 depressed subjects reported that their symptoms were “much improved” or “very much improved”
(17) .
Despite the predominance of depression in bipolar disorder
(18), studies of drug treatments for bipolar depression are far and few between compared with those for treatment of acute mania and for bipolar maintenance treatment. At present, an olanzapine-fluoxetine combination
(19) and quetiapine
(20) are the only FDA-approved treatments for bipolar depression. Although there have been placebo-controlled studies of monotherapy with lithium
(21), lamotrigine
(22,
23), and divalproex sodium
(24,
25) and controlled studies of several adjunctive antidepressants
(21,
26 –
30) and pramipexole
(31,
32), there remains an urgent need to evaluate potential therapeutic treatments and augmenting agents. This study was conducted to assess the efficacy and safety of adjunctive modafinil in the treatment of bipolar depression.
Method
Study Sites, Participants, and Design
This multisite study included University of California, Los Angeles (UCLA); University of Texas Southwestern Medical Center at Dallas; University of Cincinnati; and, in Germany, University of Munich and University of Freiburg. The principal investigators at each site obtained approval from their respective institutional review boards. Quarterly reviews of study progress were conducted by the Stanley Foundation Bipolar Network Data Coordinating Center in Bethesda, Md., from time of initial recruitment until December 31, 2002; a group based at UCLA conducted data and safety monitoring from January 1, 2003, through December 31, 2004, when the study ended.
Patients with bipolar disorder were recruited from academic settings, community mental health outpatient clinics, physician referrals, and local advertisements. All participants were between the ages of 18 and 65 and provided written informed consent prior to evaluation and randomization. U.S. participants had to be able to read English. For the German sites, the protocol and consent material were translated into German.
The study was an acute-phase 6-week randomized double-blind placebo-controlled evaluation of adjunctive modafinil in patients with bipolar I or II depression that was inadequately responsive to a mood stabilizer with or without concomitant antidepressant therapy. All medications were at stable doses for at least 2 weeks prior to randomization and were held constant during the acute trial. Patients had to have a diagnosis of bipolar I or II depression, as confirmed by assessment with the Structured Clinical Interview for DSM-IV (SCID;
33 ), and moderate symptom severity as rated by a score ≥16 on the Inventory of Depressive Symptoms—Clinician Rated (IDS;
34 ). The SCID was administered by clinical research assistants who had received training under the supervision of the principal investigator at their site. Interrater reliability for the diagnosis of bipolar disorder was established with an overall kappa value of 0.92
(35) .
The main exclusion criteria were concurrent use of nefazodone (which is a cytochrome P450 inhibitor) or a monoamine oxidase inhibitor, active suicidality, active psychosis, current alcohol or other substance abuse or dependence, an unstable general medical condition, a clinically relevant baseline laboratory abnormality, ECG evidence of ischemia or ventricular hypertrophy, a history of stimulant-induced mania, or a baseline sleep pattern of <6 hours a night. Of the 120 subjects who provided written informed consent, 30 were screened out by inclusion or exclusion criteria.
Randomization, Rating Scales, and Outcome Measures
Double-blind randomization was conducted in a 1:1 block design at each participating site. Medication dosing was started at one tablet (100 mg modafinil or placebo) a day for 1 week. For weeks 2–6, the dose was increased to one tablet twice a day (total, 200 mg modafinil or placebo). In cases of early clinical response or adverse effects, the dose could be reduced. Ratings performed at baseline and every week for the 6-week study included the IDS, which is a 30-item clinical rating scale (score range=0–90) that rates severity of depression; the Young Mania Rating Scale (YMRS;
36 ); the Clinical Global Impression—Bipolar Version scale (CGI-BP;
37 ); the Epworth Sleepiness Scale
(38), which is a subjective measurement of the likelihood of falling asleep during eight normal daily situations, each scored on a 4-point scale from “would never” to “a high chance” (a score ≥10 is considered abnormal); and the Fatigue Severity Scale
(39), which is a subjective measurement of fatigue severity (a score ≥4 is considered abnormal).
The primary outcome measure was baseline-to-endpoint change in IDS score. Rating scale scores on the last study visit served as the final data point. Although the Montgomery-Åsberg Depression Rating Scale and, to a lesser extent, the Hamilton Depression Rating Scale have been more commonly used in controlled trials for bipolar depression, the IDS was recently used as a primary outcome measure in one of the largest controlled comparison trials to date of second-generation antidepressants in bipolar depression
(40) . The IDS has been shown to be reliable and to correlate highly with the Hamilton (r=0.92; 32) and Montgomery-Åsberg scales (r=0.81; 41), and it was found to be more sensitive to change than the Montgomery-Åsberg scale
(41) . Although this greater sensitivity was driven primarily by the number of items in the instrument and not the item content, the authors suggested that this may be advantageous in clinical drug trials of antidepressants.
Secondary outcome measures included clinical response, defined as a 50% reduction in IDS score at the end of the 6-week trial; remission, defined as a final IDS score <12; baseline-to-endpoint change in CGI-BP depression severity score; and baseline-to-endpoint change on a subset of four IDS questions specifically assessing fatigue and energy-related symptoms (item 4, hypersomnia; item 20, energy level; item 23, cognitive slowing; and item 30, leaden paralysis; response on this measure was defined as a 50% reduction in score). Treatment-emergent hypomania or mania was defined as a YMRS score >13 at any time during the 6-week study. Participants were withdrawn from the trial if they experienced mood destabilization, which was defined as a CGI-BP improvement score of 6 or 7 (i.e., much or very much worse) for mania or depression on any visit relative to the baseline visit.
Data Analysis and Power
Ninety participants were randomly assigned to either the adjunctive modafinil group or the placebo group. Three patients who did not meet inclusion or exclusion criteria yet inadvertently received a randomization number were excluded (none of the three received study drug). Two patients dropped out of the study after the baseline randomization visit and before the first treatment visit. All analyses are intent-to-treat efficacy analyses performed with data on participants who received at least one dose of modafinil or placebo and one set of ratings after randomization (N=41 for the modafinil group, N=44 for the placebo group).
At the time of study design, we looked to other current controlled studies in bipolar depression to estimate the power of this study. A study performed by members of our group had a 23% placebo response and a 52% treatment response for bipolar depression
(22) . Assuming a similar effect size and a larger sample size, with a continuous measure and a two-tailed alpha of 0.05, the power in this study was 0.80.
Baseline demographic variables, response and remission rates, and percentage of participants with treatment-emergent hypomania or mania were analyzed by t tests and chi square tests. We used analysis of covariance (ANCOVA) to analyze depression severity at endpoint, as measured by the IDS, using baseline severity as the only covariate. To assess the potential contribution of concomitant antidepressant therapy on treatment response, a supplementary 22 ANCOVA was run using active medication versus placebo and presence or absence of concomitant antidepressant therapy as design factors. To evaluate the time course of antidepressant response, we used a mixed-model repeated-measures analysis across the 6-week trial, controlling for baseline IDS score. We specified an unstructured covariance matrix because it produced a better fit than compound symmetry (χ 2 =45.9, df=19, p=0.005) or first-degree autoregressive (χ 2 =36, df=19, p=0.01) models.
Results
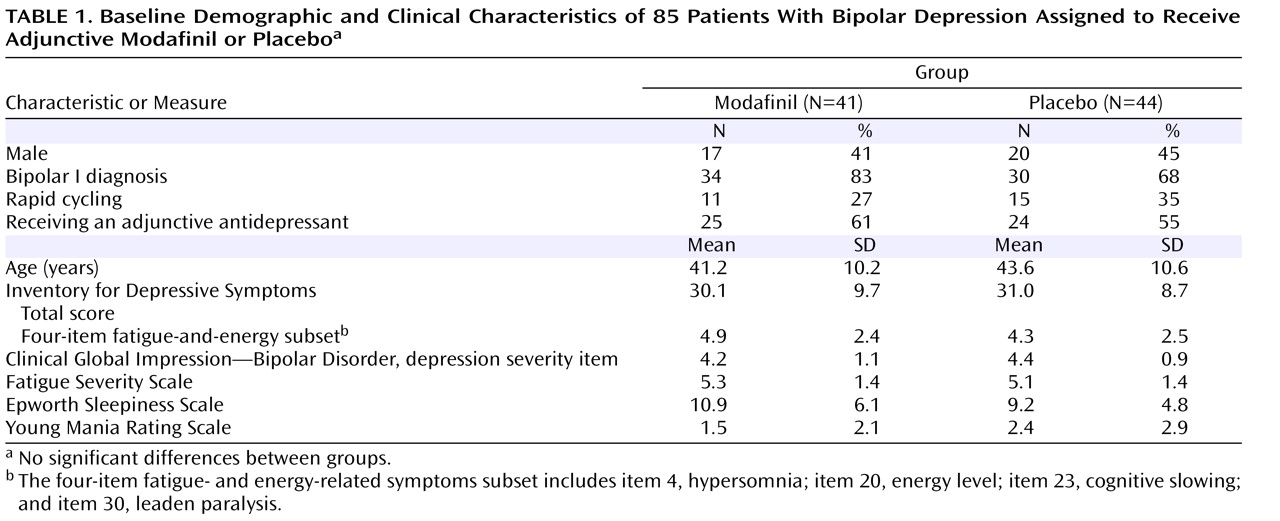
Table 1 presents the baseline demographic and clinical characteristics of the 85 participants. There were no significant differences between the modafinil and placebo groups in any of these variables.
The mean number of psychotropic medications participants were taking at the time of randomization was not significantly different between groups (modafinil group: mean=3.53 [SD=2.8]; placebo group: mean=2.88 [SD=2.0]). There was no significant difference between the modafinil and placebo groups in the percentage of participants taking lithium (modafinil group: N=12, mean dose=985.4 mg [SD=256]; placebo group: N=18, mean dose=966.7 mg [SD=322]), divalproex sodium (modafinil group: N=14, mean dose=1121 mg [SD=412]; placebo group: N=11, mean dose=1241 mg [SD=609]), lamotrigine (modafinil group: N=4, mean dose=212.5 mg [SD=165]; placebo group: N=8, mean dose=275 mg [SD=119.5]), carbamazepine (modafinil group: N=4, mean dose=875 mg [SD=250]; placebo group: N=3, mean dose=700 mg [SD=265]), or atypical antipsychotics (modafinil group, N=21; placebo group, N=15). However, a greater number of patients in the modafinil group were receiving sedative-hypnotics (clonazepam, lorazepam, or zolpidem) than in the placebo group (N=19 versus N=7; χ
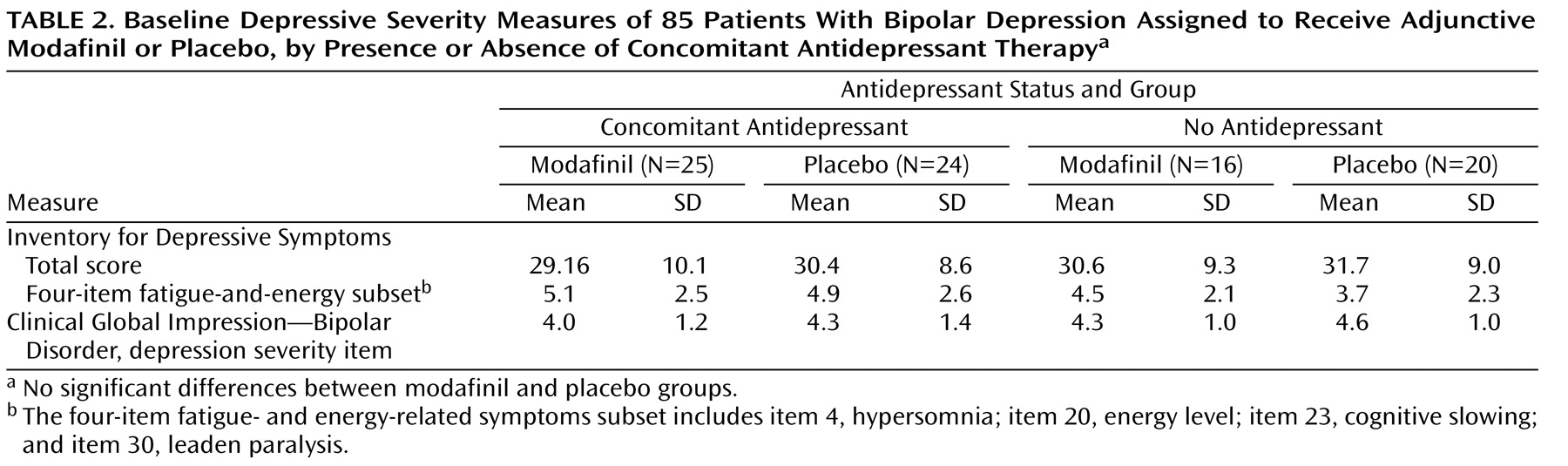
2 =9.26, p=0.002). Similar proportions of patients in the modafinil and placebo groups were receiving ongoing adjunctive antidepressant therapy (N=25 and N=24, respectively). As shown in
Table 2, there was no significant difference in mean baseline depression severity measures between those receiving and those not receiving concomitant antidepressant therapy.
A total of 58 participants (68.2%) completed the 6-week trial. The mean study drug dose for these patients was 174.2 mg for modafinil and 177.27 mg for placebo. Dropouts in the two groups (N=12 in the modafinil group and N=15 in the placebo group) did not differ significantly in the time course of dropout.
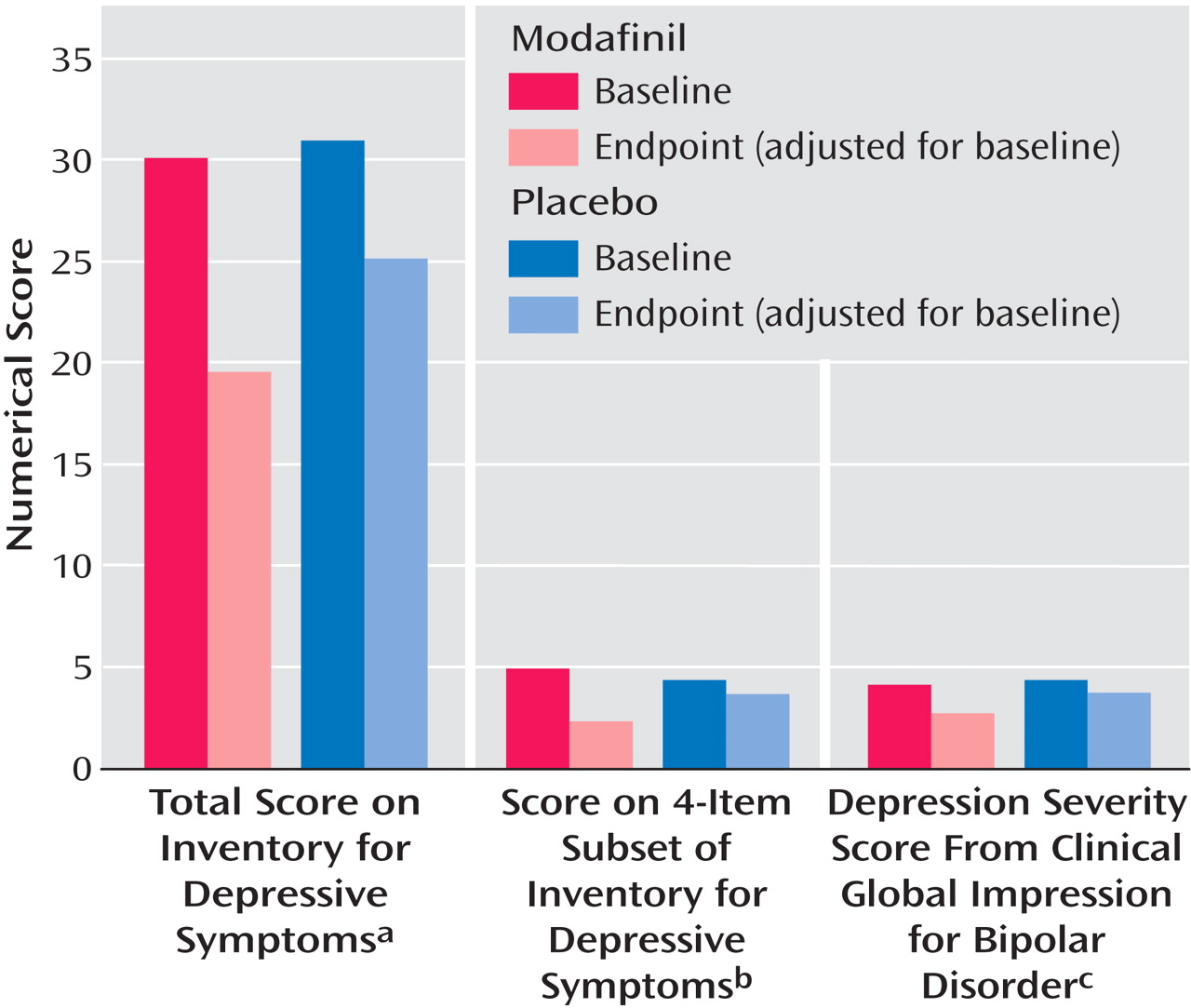
In the ANCOVA controlling for baseline scores, the endpoint scores on the IDS, four-item fatigue-and-energy subset of the IDS, and CGI-BP depression severity item were significantly reduced in the modafinil group compared with the placebo group (
Figure 1 ). The effect size was medium for each of these endpoint measures (0.47, 0.56, and 0.63, respectively). At endpoint, concomitant antidepressant therapy did not contribute to the difference between groups in IDS score (active medication: F=4.07, df=1, 80, p=0.047; antidepressant therapy: F=0.03, p=0.86; active medication by antidepressant therapy: F=0.06, p=0.81), score on the four-item subset of the IDS (active medication: F=6.48, df=1, 80, p=0.01; antidepressant therapy: F=0.02, p=0.88; active medication by antidepressant therapy: F=0.01, p=0.93), or CGI-BP depression severity score (active medication: F=7.14, df=1, 80, p=0.009; antidepressant therapy: F=0.13, p=0.72; active medication by antidepressant therapy: F=0.71, p=0.40).
The percentage of participants who achieved a 50% or greater improvement in IDS score was significantly higher in the modafinil group than in the placebo group (43.9% versus 22.7%; χ 2 =4.31, p=0.038). Differential response rates were also observed in the four-item subset of the IDS (59% versus 31%; χ 2 =6.39, p=0.01). The remission rate (final IDS score <12) was significantly higher for the modafinil group than the placebo group (39% versus 18%; χ 2 =4.55, p=0.033).
The study design did not stratify the randomization according to bipolar I versus II subtypes. Nonetheless, the endpoint IDS score, controlling for baseline score, was significantly lower in patients who had a diagnosis of bipolar I disorder compared with those who had a diagnosis of bipolar II disorder (bipolar I: N=64, baseline IDS=29.43 [SD=9.2], endpoint IDS=20.1 [SD=12.4]; bipolar II: N=21, baseline IDS=33.84 [SD=8.15], endpoint IDS=29.45 [SD=11.6]; F=6.58, df=1, 84, p=0.012). The response rate was higher in the bipolar I cohort (modafinil group: 17 of 34 [50%]; placebo group: 8 of 30 [26%]; χ 2 =3.64, p=<0.06) compared with the bipolar II cohort (modafinil group: 1 of 7 [14%]; placebo group: 2 of 14 [14%]). Similarly, the response rate (50% reduction) on the four-item subset of the IDS was higher in the bipolar I cohort (modafinil group: 23 of 34 [68%]; placebo group: 12 of 28 [42%]; χ 2 =3.84, p=0.05) compared with bipolar II cohort (modafinil group: 1 of 7 [14%]; placebo group: 1 of 14 [7%]; χ 2 =0.27, p=0.6).
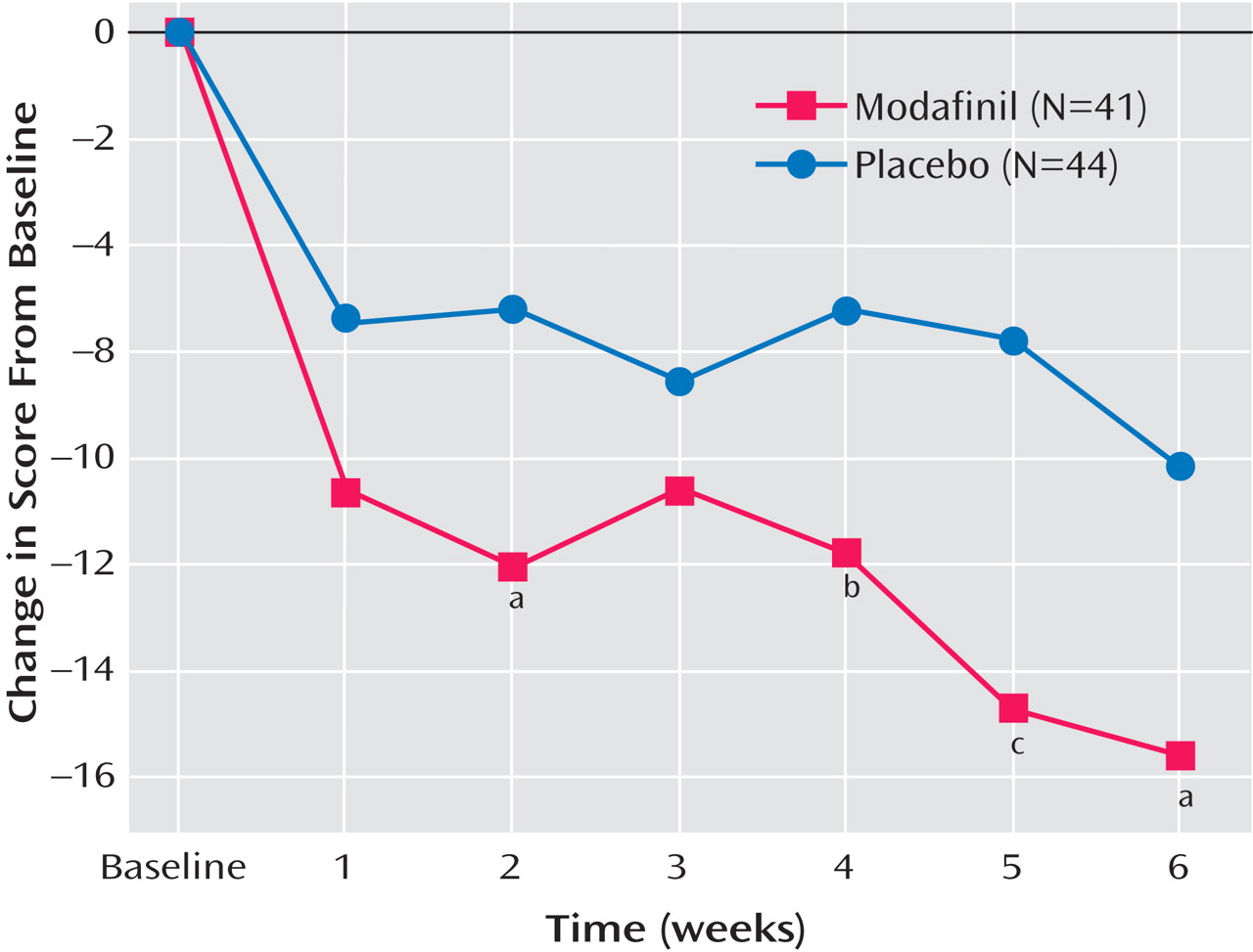
In the mixed-model repeated-measures analysis, there was a significant main effect for medication treatment (F=6.21, df=1, 82, p=0.015; see
Figure 2 ). Neither the main effect of time nor the time by medication interaction was significant, indicating a relatively consistent difference between groups over time. Patients in the modafinil group had greater improvements than those in the placebo group at every visit starting at week 2, except for week 3 (week 2: F=4.93, df=1, 82, p=0.029; week 4: F=3.42, p=0.068; week 5: F=8.26, p=0.005; and week 6: F=4.18, p=0.04).
In the ANCOVA controlling for baseline, there were no differences between groups at endpoint in YMRS score (modafinil group: mean=2.88 [SD=0.75]; placebo group: mean=3.59 [SD=0.73]), Epworth Sleepiness Scale score (modafinil group: mean=6.39 (SD=0.74); placebo group: mean=8.16 [SD=0.72]), or Fatigue Severity Scale score (modafinil group: mean=4.27 [SD=0.22]; placebo group: mean=4.5 [SD=0.21]). Treatment-emergent hypomania or mania (defined as a YMRS score >13) did not differ between the modafinil group (6 of 41 [14.6%]) and the placebo group (5 of 44 [11.4%]). There was no significant difference in treatment-emergent hypomania or mania between treatment groups by whether patients received concomitant antidepressant treatment (modafinil group: 3 of 25 [12%]; placebo group: 4 of 24 [16.7%]) or did not receive concomitant antidepressant treatment (modafinil group: 3 of 16 [18.8%]; placebo group: 1 of 20 [5%]).
There were no significant baseline or endpoint differences in blood pressure, heart rate, or weight between groups. The most common side effect was headache (four patients in the modafinil group, one in the placebo group). Other adverse events reported were hypomania as indicated by mild manic severity on the CGI-BP (one in the modafinil group, four in the placebo group), nausea (one in each group), infection (one in the placebo group), dyspepsia (two in the placebo group), insomnia (two in the modafinil group), rapid heart rate (one in the modafinil group). During the course of the study, there were two hospitalizations for mania (one in each group), one hospitalization for depression (in the modafinil group), and one depression exacerbation (in the placebo group).
Discussion
To our knowledge, this is the first placebo-controlled trial of adjunctive modafinil in bipolar disorder and one of a few studies of novel agents for the treatment of bipolar depression
(31,
32) . The results suggest that adjunctive modafinil at doses of 100–200 mg a day may improve depressive symptoms in bipolar disorder. Scores on the IDS, the four-item fatigue-and-energy subset of the IDS, and the CGI-BP depression severity item all suggest that adjunctive modafinil has some benefit in the treatment of bipolar depression. The time course of response is roughly similar to those seen in trials of quetiapine
(20), an olanzapine-fluoxetine combination
(19), and lamotrigine
(23) . Secondary analyses suggested that the presence of concomitant antidepressants did not contribute to the primary antidepressant effect of modafinil or add liability to mood destabilization.
One of the largest comparative studies to date of adjunctive second-generation antidepressants in bipolar disorder
(40) reported response rates similar to those we report here. In that 10-week trial, in which 174 bipolar depressed patients were randomly assigned to receive adjunctive sertraline, bupropion, or venlafaxine, there was no significant between-groups difference in response rates (41% for sertraline, 33% for bupropion, and 37% for venlafaxine). While the study we report here is smaller, it was placebo controlled, and the 44% response rate was similar in magnitude to rates in the antidepressant study and greater than placebo in the present study.
Although an antidepressant effect was observed for modafinil, no reduction in sleepiness, as measured by the Epworth Sleepiness Scale, or in fatigue, as measured by the Fatigue Severity Scale, was observed. It is not entirely clear whether we underdosed modafinil such that we failed to identify a change in energy or wakefulness or whether the instruments we used were insensitive in the context of patients with bipolar depression who did not have a primary sleep disorder diagnosis. Modafinil does have a dose-dependent neurobehavioral effect by which higher doses are associated with a greater degree of wakefulness
(1) . Moreover, there are data to suggest differential neuroanatomical and neurobehavioral effects of low versus high doses of modafinil. Lower doses (100–200 mg) have been shown to have a reliable effect on cognition in healthy volunteers
(42) and patients with schizophrenia
(43) and have been shown to activate the anterior cingulate
(44) . Higher doses (200–400 mg), as used in the registrational sleep disorder trials, have been shown to promote wakefulness and attention
(1,
2) and to activate anterior hypothalamic brain regions
(45) . With a mean dose under 200 mg in this study, it is possible that we underdosed modafinil and may have missed a “wakefulness” signal, although our dosing appears to have been sufficient for an antidepressant effect. We do not yet understand the potential antidepressant action of modafinil or the impact of increased attention and wakefulness on an antidepressant response. In any case, the Fatigue Severity Scale and Epworth Sleepiness Scale are relatively insensitive instruments, and they have been validated only in neurological conditions such as multiple sclerosis and systemic lupus erythematosus (Fatigue Severity Scale) or in sleep disorders (Epworth Sleepiness Scale). Neither instrument has shown a placebo-drug difference as a primary or secondary outcome measure in any non-sleep related psychiatric research.
Because these fatigue and sleepiness scales have not been validated in mood research, we planned a priori in our design to use the four fatigue- and energy-related items from the IDS—the items on hypersomnia, energy level, cognitive slowing, and leaden paralysis—which we thought were related to the neurobehavioral effect of modafinil. There was no difference between groups in this subset of the IDS at baseline, and the baseline-to-endpoint change was greater for patients in the modafinil group; moreover, this improvement was not related to the presence of adjunctive antidepressant treatment. This result would suggest that modafinil did indeed have neurobehavioral effects, but, as noted above, our data do not help delineate how these effects may have been secondary to an antidepressant effect of modafinil versus a primary impact of cognition and energy enhancement.
When considering antidepressant treatment for bipolar depression, the risk of treatment-emergent mania or hypomania must be carefully weighed against the potential benefit of the antidepressant medication. Recent data on acute treatment of bipolar depression would suggest that the placebo or course-of-illness switch rate may be as low as 4%–5%
(19,
20) . Hence, it is valuable to assess any potential increased risk associated with adjunctive antidepressant treatment. In a meta-analysis, Gijsman et al.
(46) found comparable efficacy but higher switch rates for tricyclic antidepressants compared with the newer antidepressants in trials of acute treatment of bipolar depression. The noradrenergic properties of tricyclics and of venlafaxine (which has a higher switch rate than paroxetine
[26] or than sertraline or bupropion
[40] ) may be associated with a higher risk of treatment-emergent switching. There are no known noradrenergic properties of modafinil. We evaluated mood destabilization in several ways, including manic serious adverse events, hypomanic adverse events as defined by CGI-BP criteria, and YMRS criteria. Our switch rates of 14.6% for modafinil and 11.4% for placebo were higher than in the recent placebo-controlled studies of quetiapine
(20) and the olanzapine-fluoxetine combination
(19) . It should be noted, however, that our percentage of rapid cyclers was higher than in one of the comparison studies
(20) and that our switch threshold was lower than in both studies (YMRS score >13 in our study versus YMRS score ≥15 in the olanzapine-fluoxetine combination study and YMRS score ≥16 for two consecutive visits in the quetiapine study). Furthermore, adjunctive antidepressant therapy, like adjunctive modafinil treatment, did not pose an added risk of mood destabilization. From the standpoint of other safety parameters, adjunctive modafinil appears to be safe and well tolerated. Headache was the most common side effect, and it occurred in less than 10% of patients.
This trial had several methodological limitations. First, 58% of the participants were receiving traditional antidepressant therapy that was not standardized in terms of dosing, antidepressant class, or duration of treatment. However, there was no statistical evidence that the participants receiving antidepressants differed in baseline depressive severity from those receiving antidepressants or that antidepressant therapy had any effect on our depression outcome measures at 6 weeks. Second, participants had to be treated with mood stabilizers prior to randomization, and this treatment also was not standardized in terms of dosing or type of mood stabilizer. Although a greater proportion of participants in the modafinil group were receiving adjunctive sedative-hypnotics, it is unlikely that this contributed to a significant difference between treatment groups in antidepressant response. Nonetheless, it will be important in future controlled studies of adjunctive modafinil to standardize baseline treatments prior to randomization. Moreover, a fixed-dose design may prove to be useful in assessing the effects of high doses (300–400 mg) versus low doses (100–200 mg) of modafinil on wakefulness, cognition enhancement, antidepressant response, and potential manic liability.
Our results in this 6-week trial suggest that adjunctive modafinil at doses of 100–200 mg a day may improve symptoms of bipolar depression without mood destabilization. It will be important in our 4-month open-trial continuation phase of this study to assess ongoing antidepressant response and stability.