Hippisley-Cox et al. (
2) conducted a study to determine the risk for six common cancers in schizophrenia patients. In a sample of 40,441 incident cases of cancer, odds ratios for cancer risk associated with schizophrenia and bipolar disorder were calculated with adjustments made for smoking, body mass index, socioeconomic status, comorbid physical conditions, and medication use. Analyses revealed a 47% lower risk for respiratory cancer in schizophrenia patients relative to patients without schizophrenia. In contrast, risk rates associated with bipolar illness in this cohort were comparable to those reported in nonpsychiatric samples, suggesting a relatively specific effect in schizophrenia. These findings are consistent with a recent meta-analysis of cancer incidence in schizophrenia patients and their first-degree relatives, which reported lower rates for a number of non-smoking-related cancers in schizophrenia patients as well as a lower risk for lung cancer after controlling for smoking prevalence (
1). Taken together, these studies suggest that the discrepancy between cancer risk exposure and cancer incidence in schizophrenia is consistent with a protective effect; however, the nature of the proposed protective effect is unknown.
It has been hypothesized that decreased cancer susceptibility in schizophrenia is either related to medications used to treat the illness (
3) or genetically influenced, linked with tumor suppressor genes (
4) or enhanced natural killer cell activity (
5). To date, results are inconsistent with regard to the potentially protective effects of antipsychotic medication, with some studies reporting decreased cancer rates in patients treated with antipsychotics (
6) while others have reported an increased incidence of certain cancers (i.e., colon) in patients treated with antipsychotics compared with those not treated with these agents (
2). A genetic influence on the paradoxical cancer-schizophrenia relationship is supported by family studies, in which decreased cancer rates have been reported in unaffected family members of schizophrenia patients never exposed to antipsychotic medications (
1,
7–
9), as well as case-control studies describing associations between schizophrenia and the tumor suppressor genes adenomatous polyposis coli (APC) (
10) and p53 (
11,
12). Tumor suppressor genes may act to increase risk for schizophrenia by disruption of cell growth or through abnormal apoptosis during neurodevelopment while simultaneously acting to decrease risk for cancer due to the same—in this case advantageous—apoptotic mechanism (
4).
While previous studies have focused on tumor suppressor genes and their potential role in disorders such as schizophrenia, one widely studied cancer-related gene is the
MET proto-oncogene, whose primary functions are related to the metastasis of several forms of cancer and peripheral organ development and repair (
13–
16).
MET spans 125 kb on chromosome 7q31, consists of 21 exons, and codes for the MET receptor tyrosine kinase. While activation of the MET signaling pathway can result in the abnormal growth and spread of cancerous tumors,
MET also plays a critical role in cortical and cerebellar development (
17–
20), making it a candidate gene of interest for a broad range of neuropsychiatric disorders with a neurodevelopmental etiology, including autism. Campbell et al. (
21) reported that the C allele at a single nucleotide polymorphism (SNP; rs1858830) in the promoter region of
MET was significantly overtransmitted to affected family members in two independent family cohorts ascertained for autism spectrum disorders (
21) and subsequently replicated this finding in a third independent sample (
22).
Although DSM-IV-TR criteria exclude the presence of schizophrenia and autism in the same individual (
23), both disorders may be related to abnormal neurodevelopment. Although the two illnesses are clearly distinct, they have a number of clinical features in common. Autism spectrum disorders are characterized by social deficits, abnormal language development, restricted interests, and repetitive behaviors. In addition, approximately 30% of individuals with autism are significantly cognitively impaired, although the degree of impairment varies by disorder subtype (
24). Similarly, schizophrenia patients also commonly display social deficits (asociality), anhedonia (
23), and significant neurocognitive impairment (
25). Finally, data from family and molecular genetic studies suggest an overlap between these illnesses (
26).
Results
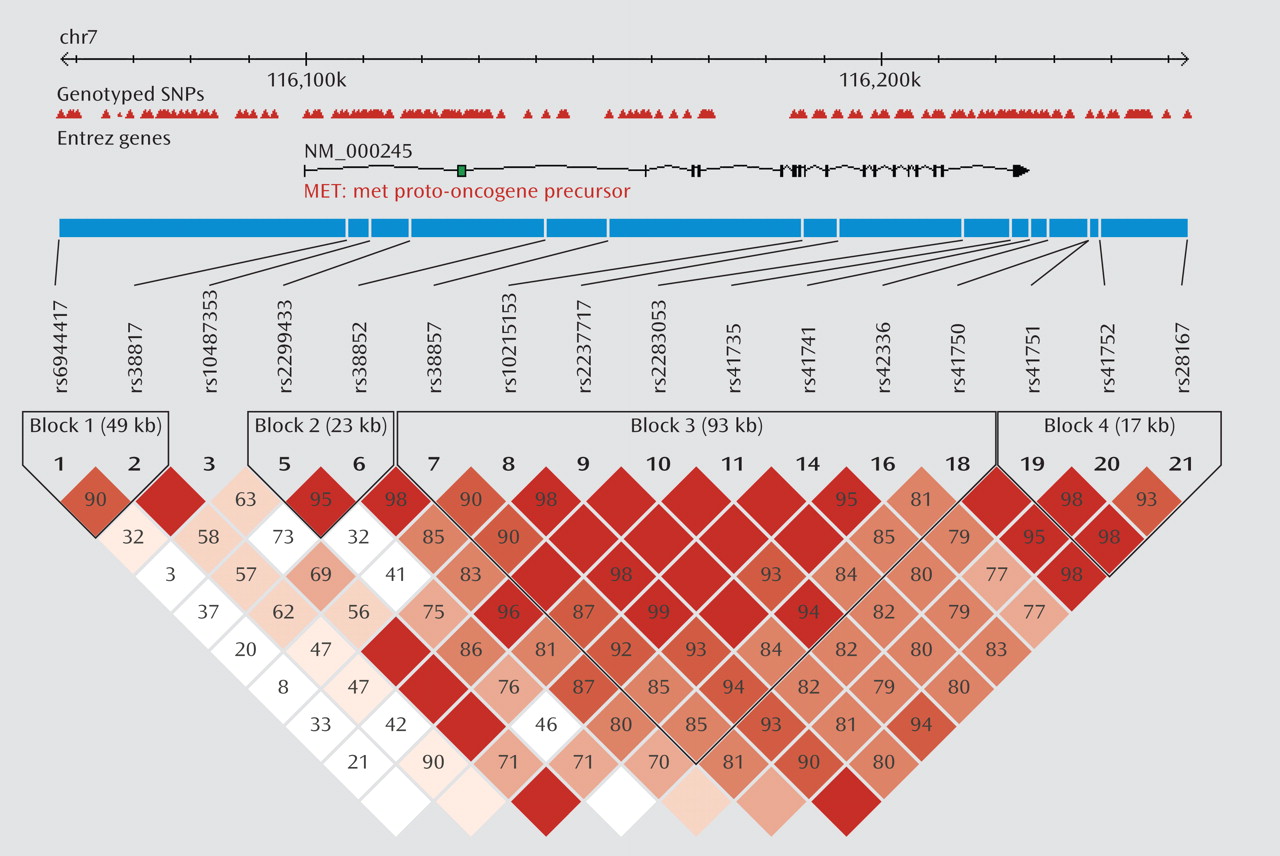
We first tested for association between
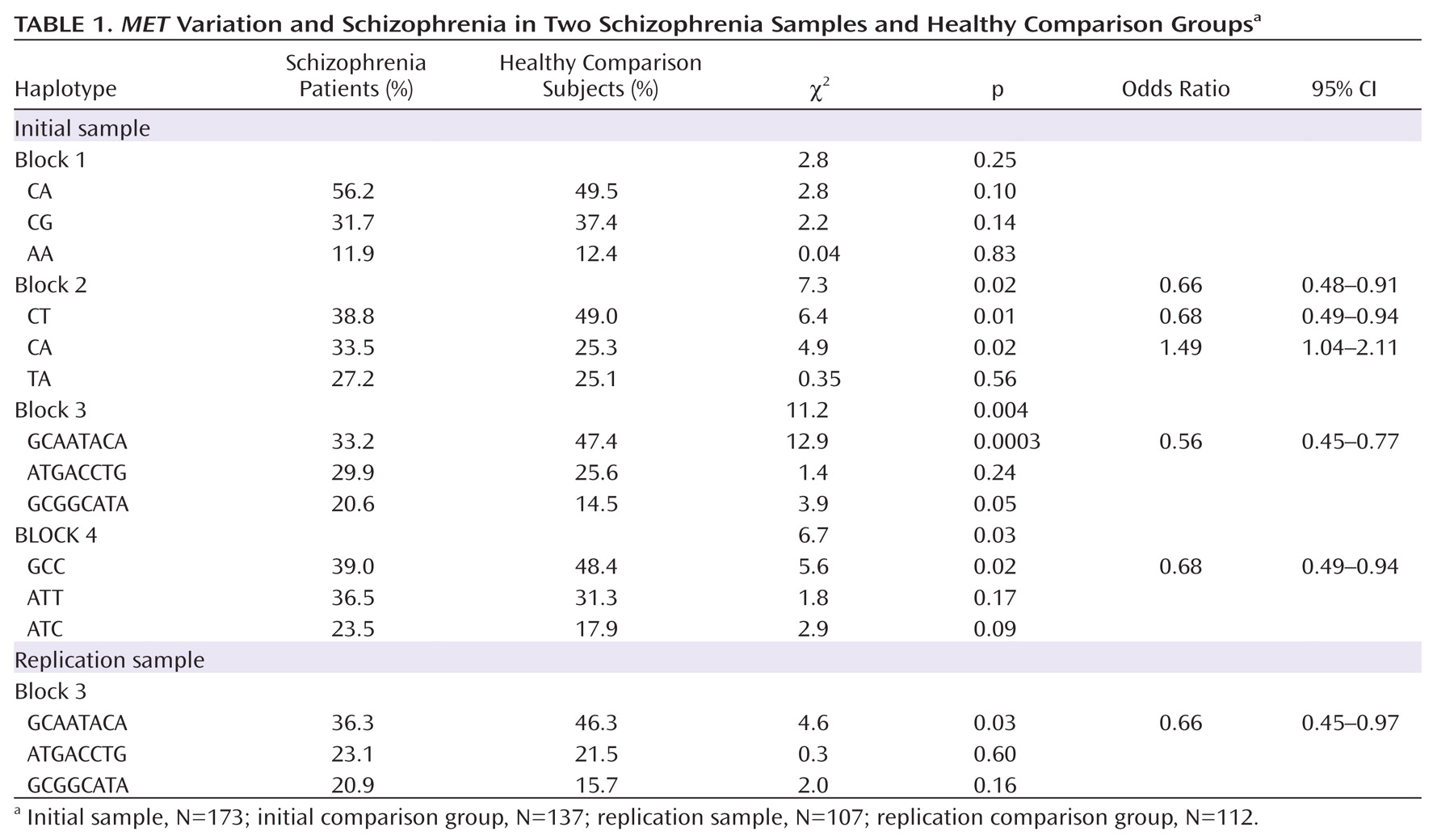
MET variation and schizophrenia in the initial sample. Four haplotype blocks were identified, each consisting of three major haplotypes. The strongest association was observed in block 3, with the most common haplotype (GCAATACA) overrepresented in the comparison group (47% frequency) compared with the schizophrenia group (33% frequency) (
Table 1). Post hoc analyses indicated that the best fitting model was a dominant model of inheritance, with individuals carrying at least one copy of GCAATACA being significantly less likely to develop schizophrenia as compared with those carrying no copies (χ
2=13.4, p=2.5×10
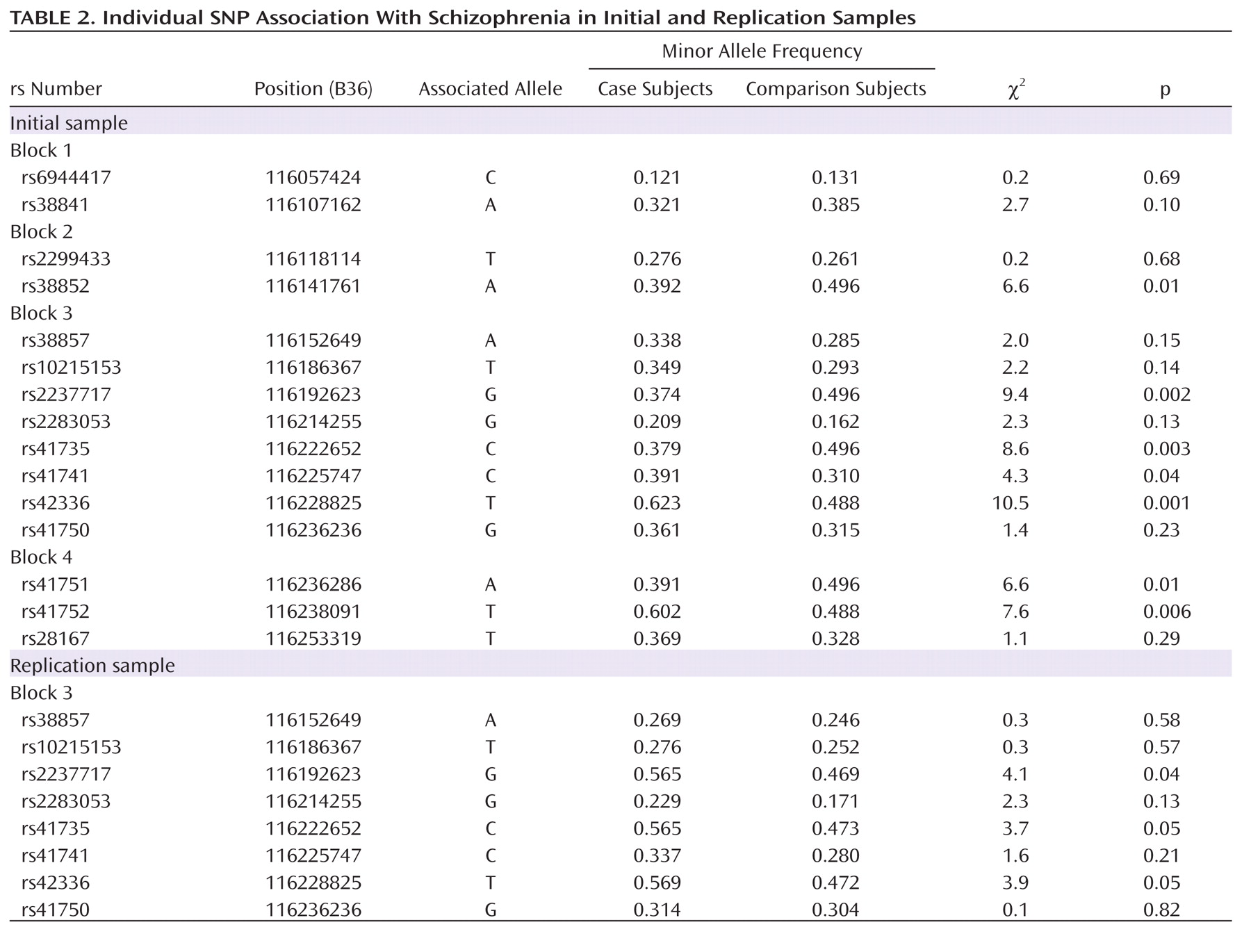
–4; odds ratio=0.40; 95% CI=0.24–0.65). Results remained significant after correction with permutation (corrected permutation p=0.0019). Individual SNP associations for the initial sample are presented in Table 2. Three of the SNPs survived correction, all residing in block 3; rs2237717 is located in intron 11, rs41735 is located in intron 19, and rs42336 is just downstream from the
MET coding region. At each of these individual SNPs, the associated "protective" allele is the ancestral allele.
For replication, we repeated the block 3 haplotype association analyses in our second independent sample and tested the effect of
MET GCAATACA on disease susceptibility. The results were highly consistent with the initial analyses, at both the haplotype and SNP levels. The most common haplotype in block 3 (GCAATACA) was overrepresented in the comparison group (frequency, 46%) relative to the schizophrenia group (frequency, 36%) (Table 1). The dominant model was also significant, with individuals carrying at least one copy of GCAATACA being significantly less likely to develop schizophrenia relative to those carrying no copies (χ
2=4.0, p=0.05; odds ratio=0.55, 95% CI=0.30–0.99). SNP associations for the replication sample are consistent with results from the initial sample (
Table 2). To address the possible influence of population stratification, we carried out a backward stepwise logistic regression with subject type as the dependent variable and GCAATACA haplotype status, as well as the second and third principal components from the population structure analysis, as independent factors. We found that although both principal components remained in the model (p≤0.001), GCAATACA haplotype remained significant (p=0.001) (SA1).
MET Association With Cognition
We next tested for an effect of MET on neurocognition. To maximize power, we merged the initial and replication data sets for all participants for whom complete neurocognitive data were available (191 schizophrenia patients and 188 comparison subjects) and tested for effects in each diagnostic group separately.
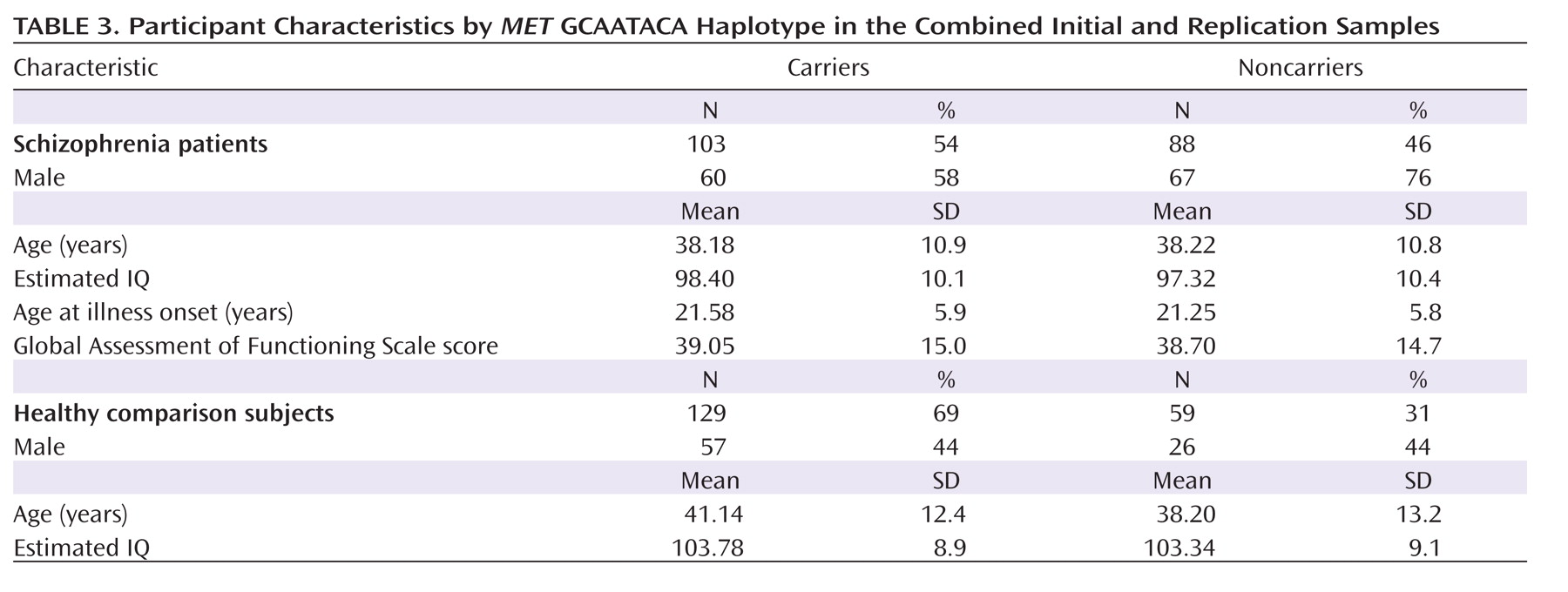
The sample characteristics are presented in
Table 3 by
MET GCAATACA haplotype group. The haplotype groups did not differ significantly in age in the comparison group or in the schizophrenia group, nor did they differ in estimated premorbid IQ. Significant differences were noted for sex distribution in the schizophrenia group (χ
2=6.8, df=1, p=0.01) but not in the comparison group. In the schizophrenia group, illness characteristics (GAF score, age at illness onset, and duration of illness) did not differ by genotype.
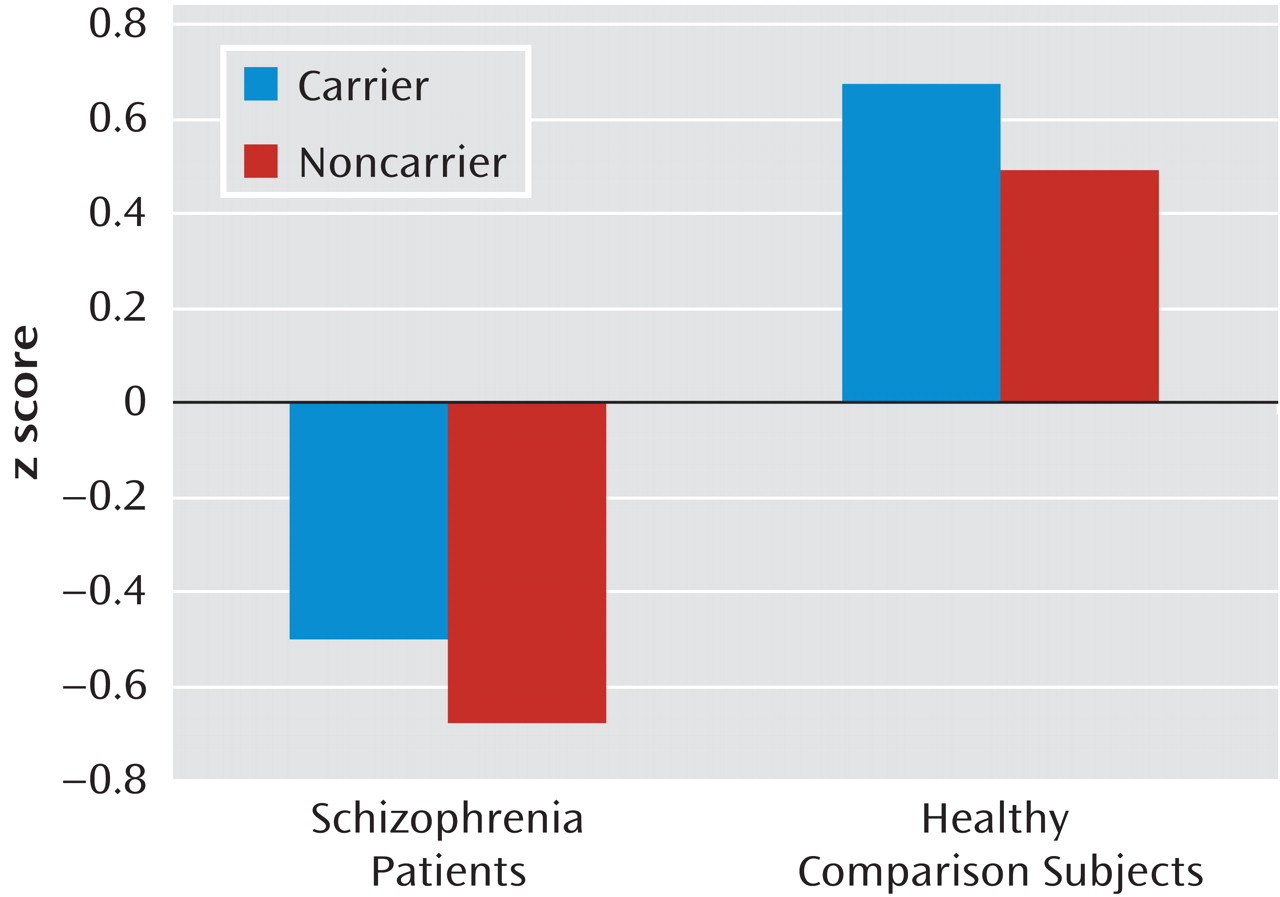
Univariate analyses of covariance in the combined comparison group revealed a significant effect of
MET GCAATACA (F=3.99, df=1, 187, p=0.05) and age (F=16.99, df=1, 187, p<0.001) on general cognitive ability (g) (
Figure 2).
MET GCAATACA carriers had significantly better cognitive performance than noncarriers, with
MET haplotype explaining approximately 2.1% of the variance in g. In the schizophrenia group, a similar pattern of performance was revealed, with carriers outperforming noncarriers (Figure 2), although the results did not reach statistical significance (
MET GCAATACA, F=1.48, df=1, 190, p=0.23; ŋ
2=0.01).
Discussion
We report a significant influence of genetic variation within the
MET proto-oncogene and susceptibility to schizophrenia in two independent cohorts. The strongest association we observed was with a haplotype spanning the majority of the coding region of
MET, as participants carrying one or more copies of the most common haplotype (GCAATACA) were significantly less likely to develop schizophrenia than those carrying no copies (odds ratio=0.40; 95% CI=0.3–0.8). These data represent the first report of an association between
MET and schizophrenia and the second of an association of
MET with susceptibility to neuropsychiatric illness. Campbell et al. (
21) reported an association between a SNP within the promoter region of
MET (rs1858830) and autism and subsequently replicated this finding and expanded the association to include several other genes within the
MET pathway (
22).
We also report that the MET GCAATACA genotype had a significant impact on neurocognition, such that healthy MET GCAATACA carriers in the comparison group performed significantly better than noncarriers on an empirically derived factor of general cognitive ability (g). This preliminary evidence of a role for MET in neurocognitive function may shed light on the mechanism through which variation in MET might influence risk for both autism and schizophrenia, as the phenotype of neurocognitive impairment is common to both diseases. These data could be interpreted to indicate that the influence of MET on disease susceptibility might not be specific to risk for schizophrenia or autism. Rather, these findings may be reflective of the effects that MET may have on brain function in general. This potential explanation warrants follow-up in other clinical disorders characterized by brain morphological abnormalities, neurodevelopmental pathology, or cognitive impairment.
The role of MET in cancer has been definitively established. There are several mechanisms by which MET influences cancer development and progression: 1) overexpression of MET—the most common alteration of MET in human tumors; 2) autocrine/paracrine activation in which MET is activated by its ligand, hepatocyte growth factor, which may be abnormally produced by cancer cells; 3) hepatocyte growth factor-independent activation via transactivation by other membrane receptors; and 4)
MET structural alterations, such as missense mutations, which result in hereditary forms of cancer. Regardless of the mechanism by which MET activity is altered, increased MET activation is associated with abnormal tissue growth related to tumor development and metastasis. In contrast, MET activation plays a beneficial role during normal physiological states and is critical to normal cortical and cerebellar development (
17–
20). Disrupted MET signaling in the cerebral cortex results in a lower number of interneurons and abnormal interneuron migration from the ganglionic eminence (
17,
18). In the cerebellum, reduced MET signaling decreases proliferation of granule cells, resulting in cerebellar volume reductions, especially in the vermis (
19). Abnormalities that result from perturbation of this system are consistent with known changes in the brains of schizophrenia patients, including dysfunction and a lower number of inhibitory interneurons in the prefrontal cortex (
34) and volumetric reductions in the vermis, both of which are correlated with cognitive dysfunction (
35). Taken together, these data suggest that
MET may influence risk for disorders such as autism and schizophrenia via a decreased level of pathway activity during critical periods of neurodevelopment. This hypothesis is supported by data from Campbell et al. (
21), who found that an autism risk allele (C) at rs1858830 was associated with a reduction in MET transcription. A subsequent study by the same group demonstrated that MET mRNA expression was decreased in the postmortem brain tissue of individuals with autism and that the level of MET expression was associated with rs1858830 in healthy comparison subjects (
36). In the present study, minor alleles of multiple intragenic SNPs (such as rs42336) were associated with an elevated risk for schizophrenia; these alleles, or perfect proxies of them, have similarly been associated with reduced gene expression in prior studies (
37,
38).
These data implicating the
MET gene in schizophrenia, coupled with the autism data, are consistent with a recent study that used computational probabilistic modeling to investigate the relationship among multiple common disorders that might be likely to share genetic susceptibility, including cancer, autism, and schizophrenia. Rzhetsky et al. (
39) proposed a disease network hypothesis based on an analysis of more than 1.5 million patient records from a clinical database and tested whether a genetic variant that predisposes an individual to a given disease may increase or decrease the risk for multiple other diseases. As in many Mendelian disorders, the tested hypothesis was that complex phenotypes are probably explained by genetic variation that is shared, in either a competitive or a cooperative manner, by multiple disease phenotypes. Among 161 disorders studied, a highly connected network of pairwise correlations emerged, including a strong positive correlation between autism and schizophrenia (p=5.78×10
–11), suggesting significant genetic overlap, such that approximately 20%–75% of autism-predisposing variants were estimated also to predispose to schizophrenia. Of particular note, a strong inverse relationship between schizophrenia and breast cancer was also observed in these analyses (p=1.22×10
–10) (
39), providing support for the hypothesis that genetic variation at a single locus, such as
MET, may have both shared and divergent effects across different phenotypes.
A specific example of the potential pleiotropic effects of
MET is provided by a recent study (
40) that investigated the effect of the
MET promoter SNP rs1858830 on susceptibility to autism in 214 autism families that were characterized for gastrointestinal comorbid conditions.
MET rs1858830 was associated with an increased risk for autism and increased susceptibility to gastrointestinal disease in 118 families with at least one child with co-occurring autism and a gastrointestinal condition; however, no association with autism was observed in families with children who did not have a comorbid gastrointestinal disease. These data provide preliminary evidence that decreased activity in the MET pathway may have both central and peripheral effects, contributing to abnormal brain development (autism) as well as dysfunctional organ repair (
40).
Our study has a number of limitations. First, medications may confound analyses in samples of patients with chronic schizophrenia; however, we would not expect that medication type or dosage would differ significantly by genotype, as the severity of illness was comparable between groups in our sample. We did not have sufficient data on medication dosage to conduct a formal test of this question. Second, the genotyped markers were selected using an Affymetrix 500K chip based on genomic spacing; we were able to capture only 71% of the common allelic variation within the MET region using the 16 SNPs analyzed. The use of genome-wide association methods raises the issue as to which analyses are considered appropriate in the context of statistically nonsignificant genome-wide results. In the strictest statistical sense, analytic follow-up of results that do not meet the genome-wide threshold after correction may not be considered valid. However, we suggest that genes such as MET, with prior data suggestive of biological plausibility related to pathophysiology, also warrant follow-up, especially given the previous association with autism and the consistency of our results in two independent samples. Finally, we acknowledge that the magnitude of the effect of MET on both disease susceptibility and neurocognitive function is relatively small, which is consistent with the complexity of the genetic architecture of schizophrenia and its related phenotypes.
The data we present here highlight the importance of assessing molecular networks that may be implicated in the pathophysiology of multiple CNS disorders with overlapping phenotypes. Our results also provide evidence for at least one potential genetic locus that may explain both a cooperative polymorphism model (increasing risk for both schizophrenia and autism) and a competitive polymorphism model (increasing risk for schizophrenia and protecting against cancer) via a specific hypothesized biological mechanism.