Family and twin studies indicate a substantial role of genetic factors in the etiology of depression. However, attempts to quantify the genetic contribution to depression have led to estimates of heritability ranging from 17% to 75%, with an average of 37% (
2,
4,
5).These discrepant estimates point to a heterogeneity in depression and can be explained by several features associated with heritability. First, cases of depression ascertained through clinical contact tend to have stronger genetic contribution than cases diagnosed by lay interviewers in the general population (
4,
5). Second, recurrent depression is more heritable than a single episode (
4,
6). Third, more reliable assessment of depression is associated with high heritability, suggesting that heritability is often underestimated due to measurement error (
7–
9). These lines of evidence all indicate a strong genetic contribution to severe and recurrent depression when it is reliably diagnosed in the context of clinical practice.
While the existence of genetic vulnerability to depression is well-established, progress in the identification of its molecular basis has been slow. Functional candidate gene studies have identified few replicable associations (
10), and genome-wide linkage studies have yielded suggestive rather than conclusive results (
11). Two genome-wide association studies (GWAS) have been published to date. A population-ascertained study of unipolar depression from the Netherlands, using healthy subjects selected for low liability to depression, detected suggestive evidence for a role of genetic variants in the
PCLO gene, but replication studies were inconsistent (
12). A second genome-wide study analyzed two cohorts of recurrent depression cases from Munich and Lausanne, along with healthy comparison subjects screened to exclude depression and anxiety, but found no evidence for genetic variants associated with depression (
13). These studies indicate that the genetic liability to depression is likely to involve multiple genetic variants of weak effects.
Discussion
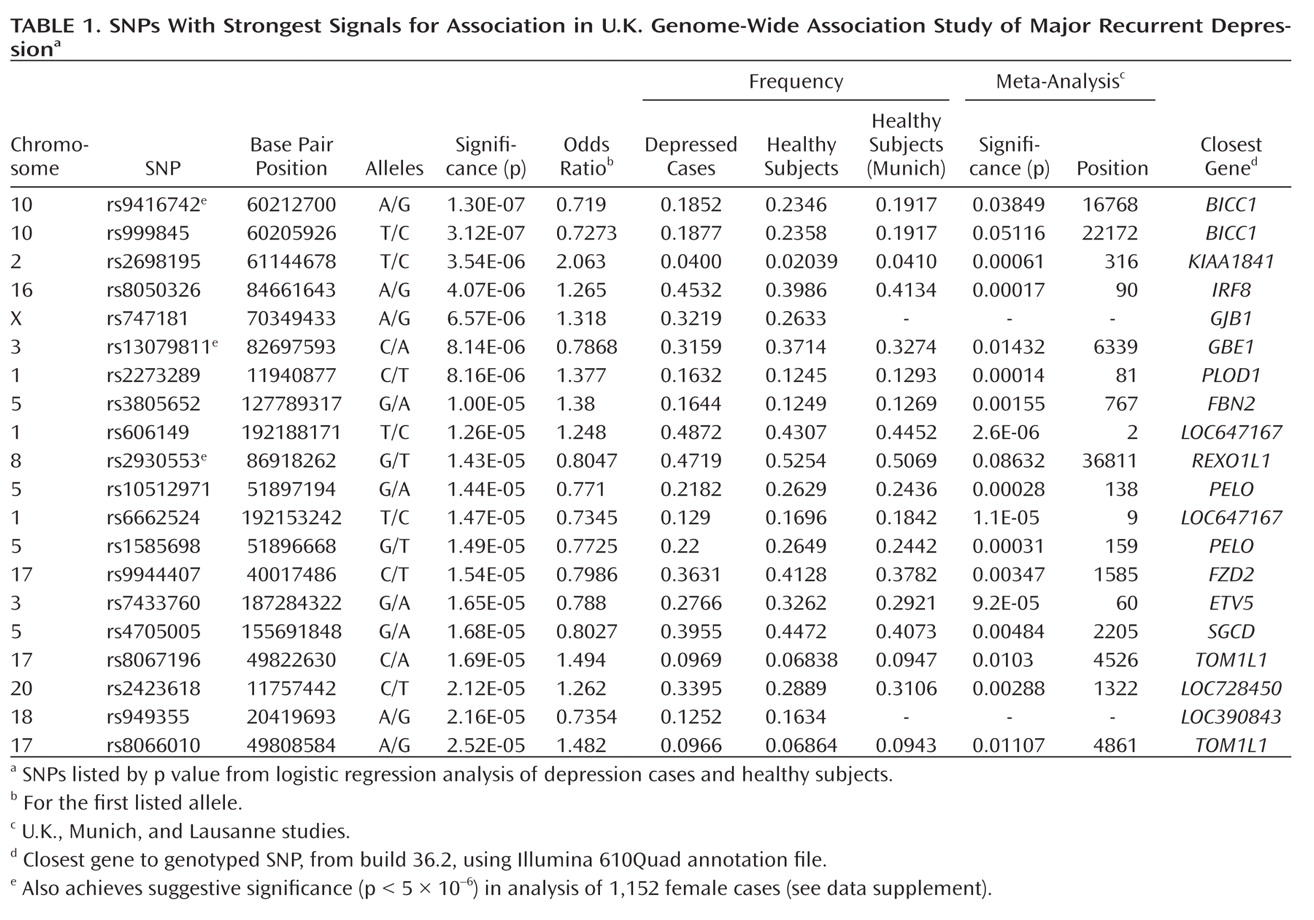
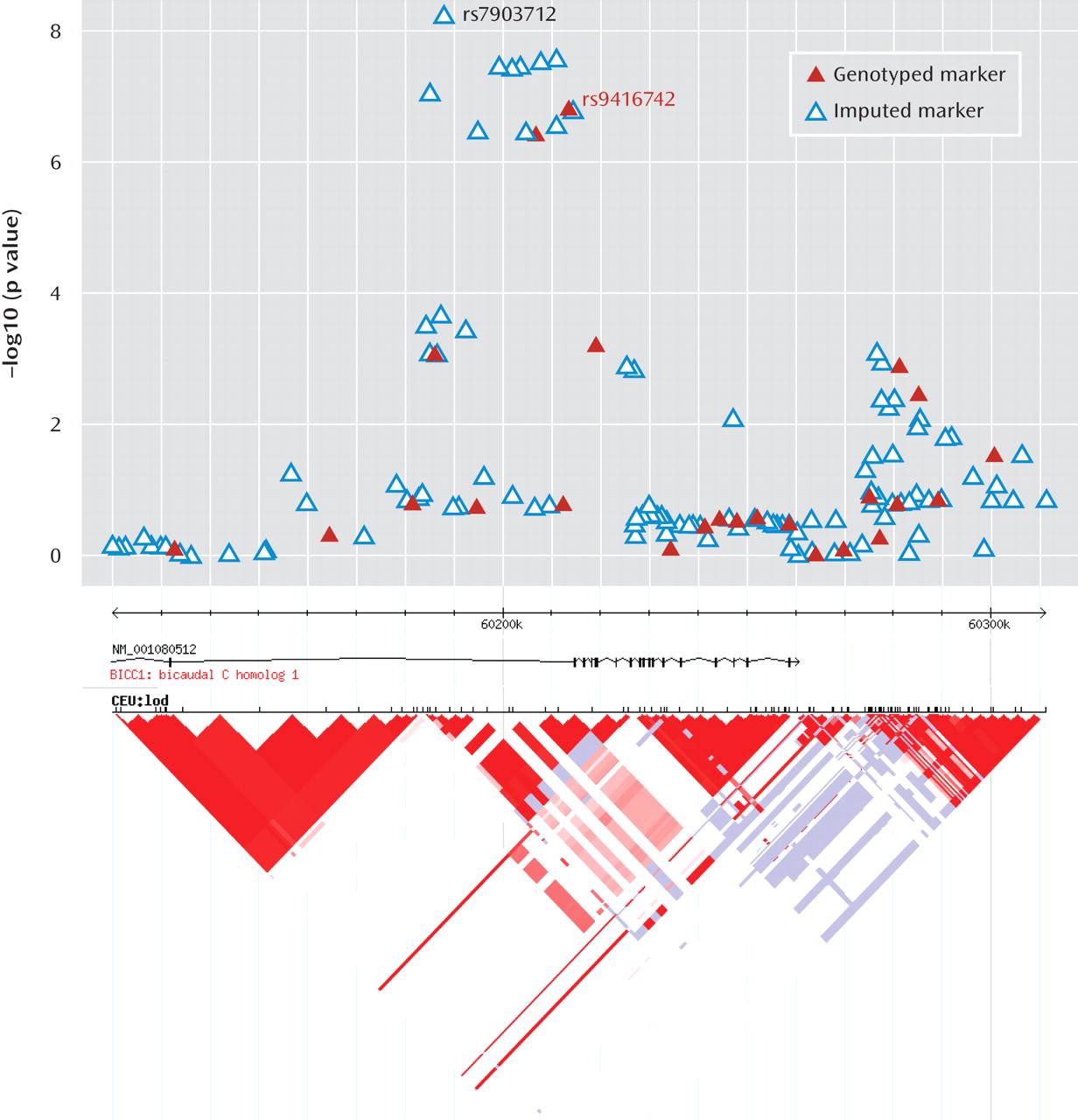
This genome-wide association study has found evidence for a role of genetic variants in the bicaudal C homologue 1 gene (BICC1) in recurrent unipolar major depression, however the finding that a minor allele of these variants has a protective effect against depression was not replicated in two independent samples of recurrent major depression collected and assessed with comparable procedures. The association of BICC1 and depression is a novel finding, with no previous published evidence for a role of this gene in neuropsychiatric disease.
The
BICC1 gene product is an RNA-binding protein which has been found to form complex interactions with RNA and with other proteins. Several independent lines of evidence suggest a role for
BICC1 in neurogenesis and plasticity, defects which may predispose to mood disorders. In
Drosophila, it is required for dorsoventral patterning of the oocyte (
32). The RNA binding activity of the protein is mediated by its KH domains (see Supplementary Figure 5) (
33), which are characteristic of the heterogeneous nuclear ribonucleoprotein k family of transcription factors and bind both DNA and RNA (
34). Other heterogeneous nuclear ribonucleoproteins are known to bind targets such as the serotonin transporter gene promoter (
35). In humans, the C-terminus of the longer isoform-1 of the protein contains a sterile alpha motif (SAM) domain (Supplementary Figure 5), thought to be a highly specific postsynaptic targeting signal (
36). This is interesting as our observed association is intronic to the long isoform, but is 15 kb 5′ from the first reported exon of a shorter isoform that does not contain a SAM domain and may highlight a haplotype which favors the expression of one or other of these isoforms. Both
BICC1 isoform RNAs are expressed widely in normal tissues and are expressed in all brain regions including cerebral cortex, hippocampus, and midbrain (see Supplementary Figures 6a-d), although
BICC1 isoform-1 RNA is more highly expressed than isoform-2, particularly in nerve tissue.
BICC1 can uncouple dishevelled-2 signaling from the canonical Wnt pathway in a SAM domain dependent manner, suggesting that the different
BICC1 isoforms may play different biological roles (
37). Gene expression studies (Array Express; http://www.ebi.ac.uk/microarray-as/ae/) suggest that
BICC1 is also upregulated by sodium valproate (a mood stabilizer and Wnt signaling inhibitor used to treat bipolar disorder), with approximately twofold increase in
BICC1 expression after valproate treatment.
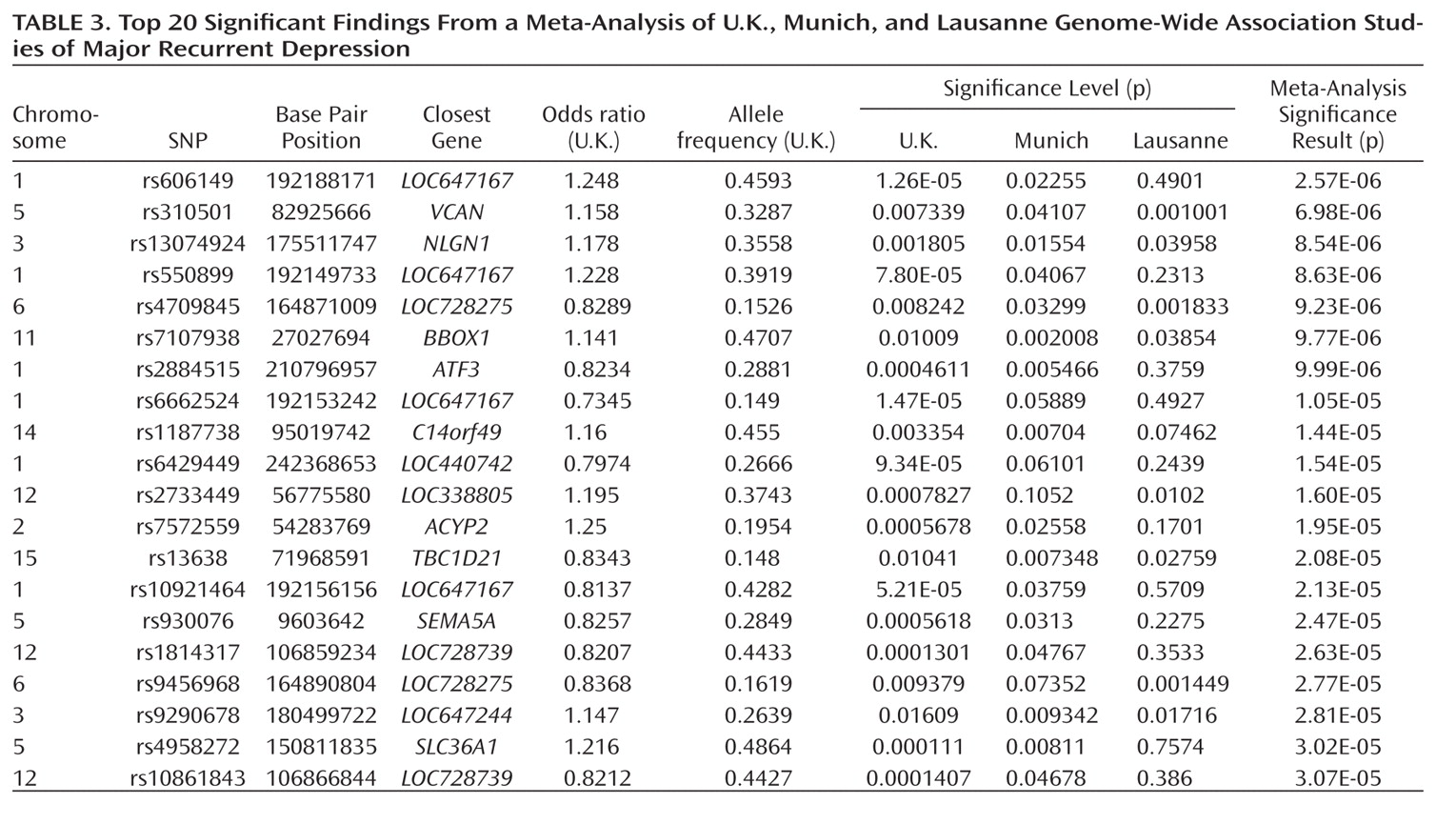
The other notable result is the meta-analysis finding of association with a SNP near neuroligin-1 (
NLGN1) on 3q26.31. Echoing our
BICC1 finding, it is known that all NLGNs are enriched at postsynaptic densities (
38).
NLGN1 encodes a neuronal cell surface protein and a splice site-specific ligand for beta-neurexins. The Ca(2+)-dependent neurexin/neuroligin complex is required for efficient neurotransmission, and involved in the formation of synaptic contacts. Neuroligins are likely to be part of the machinery employed during the formation and remodeling of CNS synapses. The neuroligin/neurexin complex has also been implicated in schizophrenia (
39), with deletions of
NLGN1 observed in autism (
40); deletions of the closely related NLGN3 and NLGN4/5 result in a variety of psychiatric phenotypes (
41,
42).
Several aspects of the finding in
BICC1 may be relevant to replication and follow-up studies. First, the strongest association was found with a SNP that was not genotyped on the Illumina panel but could be imputed with high accuracy based on genotyped markers in strong linkage disequilibrium. The high accuracy of imputation was confirmed by targeted regenotyping, and the association was statistically significant after accounting for uncertainty of imputation and for potential multiple testing of all common variants across the human genome (
27,
28). However, this effect did not replicate in either the Munich or Lausanne samples. Such nonreplication occurs frequently; with the modest effect sizes expected for genetic contributions to depression, associations will remain elusive with large variation across studies. Even for a true association, the effect size may be inflated in the discovery sample (
43,
44).
Replication of the
BICC1 finding will be necessary to determine whether this is a false positive result or a true effect which is also detectable in other studies. Such analyses are ongoing in the Psychiatric GWAS Consortium (
45), of which this study is a part. The large, well-characterized samples that form the Psychiatric GWAS Consortium will be required before we can expect to see consistent signals of association through meta- and mega-analysis. The Psychiatric GWAS Consortium analysis should help resolve the role of
BICC1 and other putative associations in depression. However, replication is likely to remain a substantial challenge for all psychiatric disorders. While large sample sizes increase the power to detect genetic risk loci, variation between studies will be unavoidable, and potentially unquantifiable. Sources of diversity between studies include those related to phenotype (e.g., criteria for depression, assessment measures of depression for both cases and comparison subjects), those related to genotype (e.g., population ancestry, genotyping panel used), and more subtle effects such as differences in patient ascertainment methods by study or cultural differences in seeking help for depression, which may result in heterogeneous case samples even though all patients meet clinical criteria for recurrent depression.
In order to evaluate the present results and to make a qualified comparison with other studies, the strengths and limitations of the study should be considered. The study used recurrent major depression, the type of depression that has been shown to be most heritable (
4,
8), with collection of cases in the U.K. by the same clinical team giving a homogeneous clinical sample. As depression is common in the general population, excluding cases of lifetime depression from the comparison sample can substantially increase power of a study. We have therefore used a sample of comparison subjects screened to exclude those with higher liability to depression. The ability of the present study to detect true associations was limited by its sample size, which was comparable to the previously published genome-wide association studies (
12,
13).
In conclusion, the present study provides evidence for a gene encoding a Wnt signaling component, BICC1, in the pathogenesis of depression. The meta-analysis implicated NLGN1, whose role in formation and remodeling of CNS synapses makes it a convincing functional candidate gene for depression. However, replication of these findings in other studies will be essential to confirm their role in depression. The results of this and other genome-wide association studies in depression suggest that effects of common genetic variants on depression are individually weak and multiple large samples will be needed to provide a more comprehensive picture of common genetic variants in depression.