Genomic microdeletions and microduplications (commonly termed copy number variants) are now known to contribute to the etiology of schizophrenia (
1–
4), autism (
5,
6), and intellectual disability (
7,
8). In schizophrenia and related psychoses, the most robust findings concern microdeletions at chromosomes 1q21.1, 15q11.2, 15q13.3, and 22q11.2 (
2–
4) as well as the
NRXN1 gene (
9) and microduplications at chromosomes 16p11.2 (
10) and 16p13.1 (
11). The finding of a nonspecific relationship between neurodevelopmental phenotypes and specific copy number variants appears to be a general feature. For example, the schizophrenia-related deletion at 1q21.1 has been reported in cases of autism (
6) and intellectual disability (
7). The same appears to be true for deletions at 15q13.3 (
8) and 15q11.2 (
12) and duplications at 16p11.2 (
6) and 16p13.1 (
13). That the same copy number variants associate with schizophrenia, autism, intellectual disability, and other neurodevelopmental disorders indicates a genetic overlap among these seemingly disparate diagnoses.
Aiming to discover novel pathogenic copy number variants, we performed a genome-wide copy number variant scan of 22 Danish schizophrenia patients with an early onset of disease (aged 10–15 years), under the premise that developmental forms of the disorder would be particularly enriched in this group (
1). One of the patients carried a 6-Mb duplication of chromosome 15q11.2-q13.1. This duplication involves the Prader-Willi syndrome (Online Mendelian Inheritance in Man [OMIM] number: 176270) and Angelman syndrome (OMIM number: 105830) critical region. These syndromes derive, respectively, from paternal and maternal chromosomal deletions (as well as other mechanisms [see Discussion section]). Duplications of the Prader-Willi/Angelman syndrome critical region were recognized in several case reports in the mid-1990s as a separate genomic syndrome, the 15q11.2-q13.1 duplication syndrome (OMIM number: 608636), which commonly includes features such as autism, intellectual disability, seizures, developmental delay, and minor dysmorphic features (macrocephaly, down-slanting palpebral fissures, expressionless face), most often when the duplications are maternally derived (
14–
16). These duplications are currently among the most consistent cytogenetic findings in autism spectrum disorders, estimated to account for 0.5%–3% of all cases (
17–
19), although the more realistic rate is closer to approximately 0.6%, based on 13 observations in the largest study, which consisted of 2,268 cases (
18).
Given the evidence that this region is important in different neurodevelopmental disorders, we investigated the involvement of chromosome 15q11-q13 duplications in schizophrenia and related psychoses in a large European sample consisting of 7,582 patients affected with schizophrenia or schizoaffective disorder and 41,370 comparison subjects. Since we did not have DNA samples for many of the identified carriers' parents, we used a methylation-sensitive Southern hybridization analysis to determine the parental origin of the duplications. This analysis distinguishes between maternally and paternally derived duplications without genotyping the parents of carriers.
Discussion
Duplications of the Prader-Willi/Angelman critical region are a recognized genomic syndrome (OMIM number: 608636), wherein the most commonly observed features include autism, developmental delay, intellectual disability, seizures, and hypotonia. In most cases where the parental origin of the duplications has been assessed, the syndrome-associated features were present only when duplications were maternally derived (
14–
16), although a developmental phenotype has been observed in single carriers of paternally derived duplications (reviewed in
19).
In the present study, we report that maternally derived 15q11-q13 duplications may also act as risk factors for schizophrenia and related psychoses. The carriers with psychosis also show known features of 15q11-q13 duplication syndrome and other genomic syndromes (e.g., developmental delay and intellectual disability) (
Table 1). However, other features, including seizures and hypotonia, were not observed in the medical records available to us for the carriers. Only one of the carriers with psychotic illness has a previous diagnosis of autism (case 2). The other carriers have no known history of autistic features, and therefore we judge it unlikely that they represent autism patients misdiagnosed as schizophrenia patients.
Although the occurrence of 15q11-q13 duplications in schizophrenia and psychotic illness is rare, and the significance level of association is nominal (p=0.01), the hypothesis for association is supported by two independent lines of evidence. The first comes from observed differences in the clinical manifestation of the various genotypes causing Prader-Willi and Angelman syndromes (
27,
28). Angelman syndrome arises from disruptions that lead to a lack of maternally expressed gene product(s), maternally derived deletions of the critical interval, paternal uniparental disomy, and mutations of the key maternally expressed imprinted gene
UBE3A. Patients with Angelman syndrome have severe to profound mental retardation, microcephaly, seizures, and ataxia and almost always lack speech (
29,
30). Prader-Willi syndrome is caused in approximately 70% of cases by deletion of the paternal chromosome for the critical region (
27). Most other cases have maternal uniparental disomy, having received two maternal copies of the region, with the paternal chromosome being absent. Rare cases are caused by mutations in the imprinting center, leading to methylation defects. Despite diverse mechanisms, all individuals with Prader-Willi syndrome have in common a lack of paternally expressed gene product(s). Prader-Willi syndrome is characterized by a failure to thrive in infancy, mild learning disabilities, and, on emerging from infancy, an abnormal satiety response to food intake and obsession with food, sexual immaturity, short stature, and a characteristic physical phenotype (
31). In addition to these core deficits, individuals with Prader-Willi syndrome are prone to affective disorders, including mood instability, nonpsychotic depression, and psychosis, features not observed in Angelman syndrome (
29,
30). Moreover, and of particular relevance to our finding, Prader-Willi syndrome resulting from maternal uniparental disomy is several-fold more frequently associated with psychotic episodes than the paternal deletion subtype (
Table 2). This has been reported in three independent studies from different countries. One study on Prader-Willi syndrome conducted in the United Kingdom identified psychotic illness at a significantly higher rate in 33 patients with maternal uniparental disomy compared with 82 patients with paternal deletions (61.8% versus 16.5%, respectively, p<0.001, odds ratio=8.2) (
27). Two other studies also reported that the maternal uniparental disomy subtype is associated with mixed symptoms of bipolar mood disorder and psychosis at high frequencies (
32,
33). Together, these studies suggest that having two maternal copies of the chromosome (leading to overexpression of maternally derived genes) is the important factor for risk of psychosis in Prader-Willi syndrome, rather than the loss of expression of genes from the paternal chromosome. This is consistent with our observation of overrepresentation of maternally derived duplication of the 15q11-q13 region in schizophrenia and schizoaffective disorder. Interestingly, among the carriers identified in our study, case patient 3 has a schizoaffective disorder, case patient 5 (schizophrenia) has prominent affective symptoms, and case patient 6 (bipolar disorder) also has prominent psychotic features (see section 3 in the data supplement), similar to the common presentations reported by both Vogels et al. (
33) and Verhoeven et al. (
34).
The second line of evidence comes from the confirmed role of 15q11-q13 duplications in the etiology of autism (
17–
19). As discussed in the introduction, there is now a strong and growing body of evidence that autism and psychotic illness partially share genetic etiology, with copy number variants that associate with schizophrenia and related psychoses also being observed at elevated rates in autism. Our current finding adds another locus to the list of copy number variants that increase susceptibility to both disorders.
The 15q11-q13 region contains a cluster of imprinted genes (genes that are expressed only when transmitted from either mother or father but not both). Several of these are only expressed from the paternal chromosome, while at least one gene,
UBE3A, is only expressed from the maternal chromosome and is robustly imprinted in the brain (
35). The product of this gene, E6-AP ubiquitin ligase, regulates the degradation of certain proteins in neurons such as the synaptic protein Arc, which promotes the internalization of AMPA receptors at excitatory synapses, thus linking
UBE3A to regulation of glutamatergic signaling and synaptic development (
35). A mouse model lacking
UBE3A shows impaired long-term potentiation in the hippocampus and, consequently, a learning and memory deficit (
36). Another gene in the region, ATP10A, encoding an amino-phospholipid-transporting ATPase involved in transporting phosphatidylserine and phosphatidylethanolamine from one side of a lipid bilayer to another, is of unknown imprinting status but is probably maternally expressed in the brain in at least some individuals (possibly depending on gender) (
37), while the mouse
ATP10A is not imprinted in the brain (
38).
Chromosome 15 harbors several loci implicated in schizophrenia and other neurodevelopmental disorders. There is a series of low copy repeat regions near the centromere. Such regions are liable to create nonallelic homologous recombination between the parental chromosomes because of the high homology between these repeats, resulting in deletions or duplications of the regions in between (
25). There are five well-recognized low copy repeat regions in the 15q11-q13 region (commonly termed BP1–BP5, reviewed in Hogart et al. [
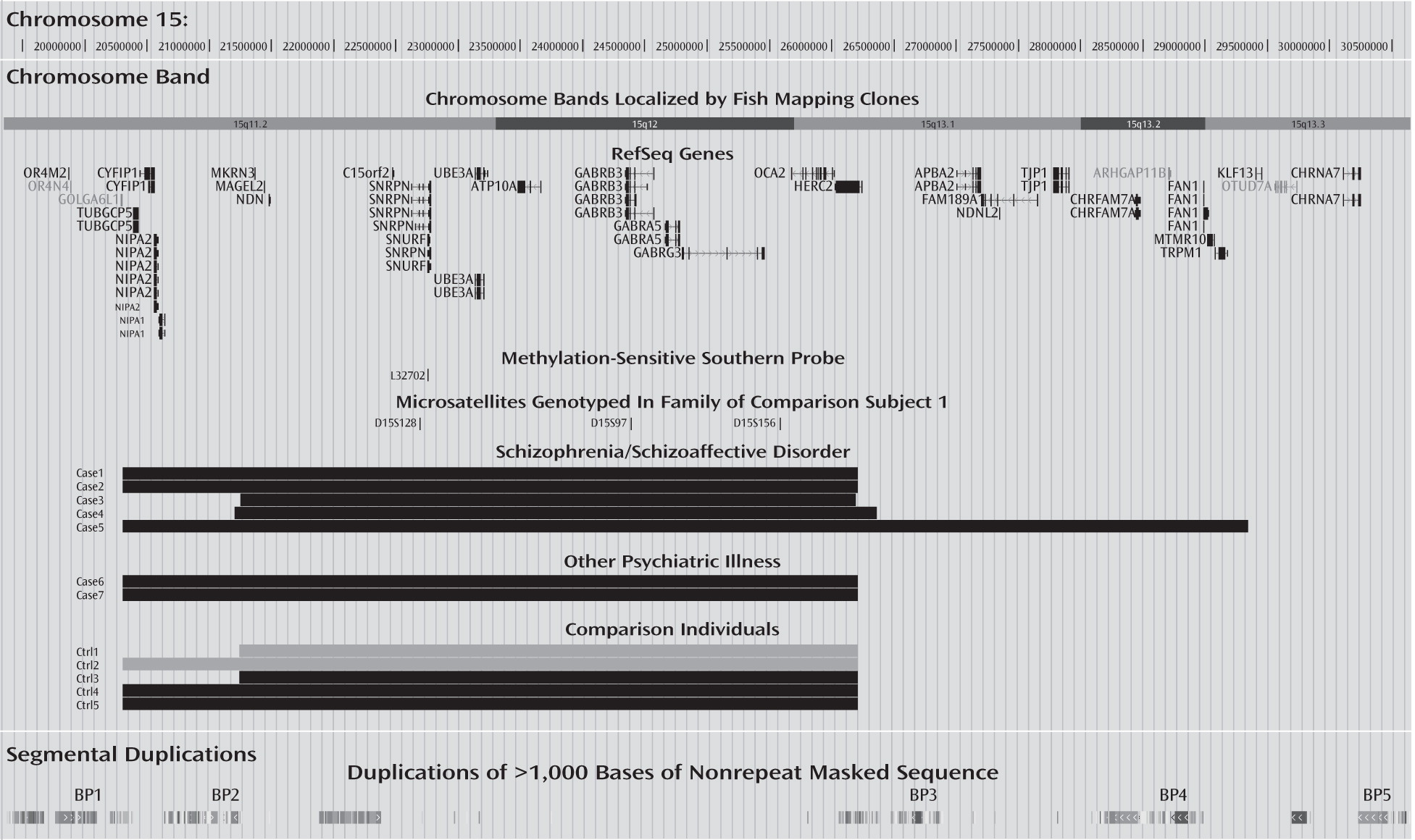
19]). Our finding differs from previous findings concerning the 15q proximal region, which have implicated deletions at 15q11.2 between BP1 and BP2 (chromosome 15: 20,306,549–20,777,695) and at 15q13.3 between BP4 and BP5 (chromosome 15: 28,723,577–30,302,218) in schizophrenia (
2–
4) as well as duplications in general occurring within the BP1–BP5 region in autism, intellectual disability, and schizophrenia (
39). The study by Itsara et al. (
39) combined data from different genome-wide copy number variant studies, and in fact the only schizophrenia case reported by these authors is that of case patient 4 in the present study (first reported by the International Schizophrenia Consortium [
3]). Our data, together with previous findings of high occurrence of psychosis in the maternal uniparental disomy subtype of Prader-Willi syndrome (
40), suggest that maternally expressed gene products in the region between BP2 and BP3 (
Figure 1) might be involved in mediating risk of psychosis. Also, our study provides new suggestive evidence of psychotic illness belonging to the phenotypic spectrum of the 15q11-q13 microduplication syndrome (OMIM number: 608636).
A limitation to our study, however, is the rarity of 15q11-q13 duplications in schizophrenia and related psychoses in general, and therefore a follow-up study of individuals with 15q11-q13 microduplication syndrome is needed to estimate the prevalence of psychotic features in adulthood in the syndrome. Another limitation is the lack of DNA samples from close family members of most of the identified carriers to determine transmission status of the duplications and cosegregation with illness. Also, we have no neuropsychiatric assessment of the carriers identified among comparison subjects. These are individuals who have participated in various genetic studies at deCODE Genetics (diagnosis is provided in
Table 1 for the two who participated as patients in a specific disease study). All five subjects were adults at the time of their recruitment, and therefore it is unlikely that they have a severe form of the duplication syndrome. Our study, together with that of Glessner et al. (
18), provides a good estimate of the prevalence of maternally derived 15q11-q13 duplications in autism and schizophrenia spectrum disorders as well as in the control population, with a prevalence of 0.5–1 out of 100, 1,000, and 10,000 individuals, respectively.
In conclusion, our findings of schizophrenia and psychotic illness in carriers of maternally derived 15q11-q13 duplications suggest that an excess of maternally expressed gene products in this region may be involved in the etiology of psychosis. Although rare in psychotic illness compared with autism spectrum disorders (0.05% versus 0.6%, respectively), exploration of the effects of this duplication in model systems has the potential to provide insight into the biological processes underlying psychosis.