Adolescence appears to be a unique period of brain development in schizophrenia (
13,
14), and changes during normal adolescence bring about hippocampal volume reduction (
15). The mechanisms underlying hippocampal volume reduction in schizophrenia are unclear and could be related to genetic, environmental, antipsychotic medication, and/or illness-related factors (
16,
17). To explore the contribution of a familial/genetic mechanism to the structural abnormalities, several studies have compared patients to their unaffected healthy siblings or other first-degree relatives (
18,
19).
The results from these studies, and thus whether a reduction in hippocampal volume represents a familial/trait marker for schizophrenia, remain unclear.
Table 1 in the present study provides an overview of studies examining hippocampal volumes in nonpsychotic relatives of adult-onset schizophrenia patients. Findings have been inconsistent, with six studies reporting smaller hippocampal volumes in first-degree relatives compared with healthy subjects, suggesting familial/genetic liability (
16,
20–
24). In contrast, five other studies failed to find any decrease in hippocampal volumes for unaffected siblings of adult patients (
25–
29) (
Table 1). Studies in ultra-high-risk individuals also address the question of whether hippocampal volume reductions could be familial/trait markers and whether the changes could take place before the onset of psychosis. In the present analysis, the studies are also inconsistent, with four showing that volume deficits do not appear until after the onset of psychotic illness (
30–
33) and three finding ultra-high-risk patients to have decreased hippocampal volume (
34–
36). It is important to note that many subjects in these studies had various psychotic symptoms and were exposed to typical or atypical antipsychotics at clinical dosages lasting from a few days to approximately 1 month, and thus the effect of antipsychotics on hippocampal volume remains a confounding factor. As a whole, these studies suggest that hippocampal volumes may be differentially affected, depending on the stage and type of psychosis, but fail to provide convincing evidence about the use of hippocampal volume as a familial/trait marker.
Childhood-onset schizophrenia, defined as onset of psychotic symptoms before age 13 years and diagnosed using unmodified DSM-IV criteria, is a rare form of the illness that is continuous with its adult counterpart (
13,
37) and shows a robust gray matter loss during adolescence that appears to be an exaggeration of the normal cortical gray matter developmental pattern (
14,
38–
41). We previously reported a moderate, nonprogressive reduction in hippocampal volume bilaterally (
13,
42). Although there are salient volume reductions in the hippocampus associated with adult-onset schizophrenia, there have been no studies examining longitudinal hippocampal volume change in either the siblings of our patients or relatives of adult-onset patients.
Discussion
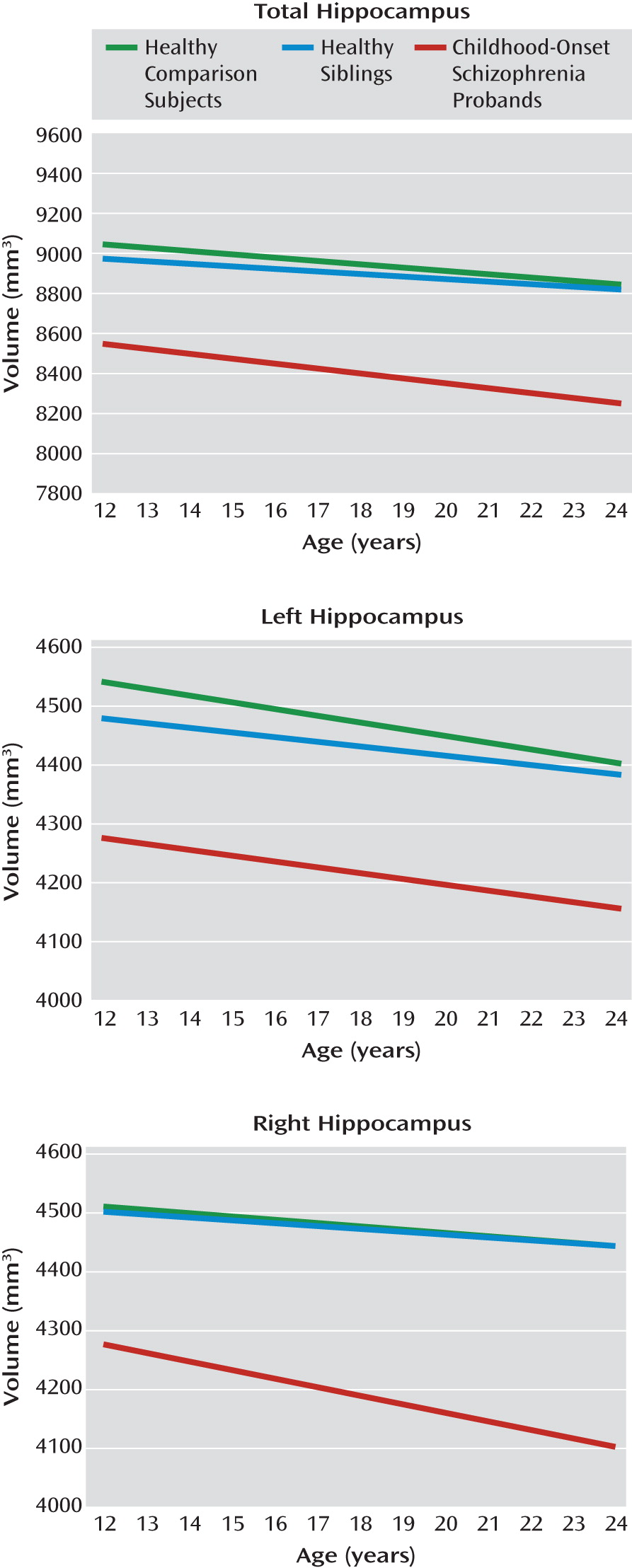
Healthy siblings of childhood-onset schizophrenia probands had no hippocampal volume deficits relative to healthy comparison subjects. However, childhood-onset schizophrenia patients showed bilateral fixed deficits in total hippocampus volumes. Similarly, the linear trajectories (slopes) across age for total, left, and right hippocampal volumes in siblings as well as childhood-onset schizophrenia probands did not differ from those of healthy comparison subjects.
These findings extend our previous reports of fixed total hippocampal volume deficits in childhood-onset schizophrenia patients (
13,
42) in a much larger sample. The volume deficits, 6%–7% at the average age, are larger than those seen in cross-sectional studies of adult schizophrenia (4%–5%) (
17,
31), which is consistent with the clinical evidence that childhood-onset schizophrenia represents a more severe phenotype of the illness. Since hippocampal volume deficits appear early in childhood-onset schizophrenia and are comparatively nonprogressive, these findings also support a static hippocampal lesion suggested by the animal models of schizophrenia (
56–
59).
Contrary to our a priori hypothesis, healthy siblings of childhood-onset schizophrenia probands showed no hippocampal volume loss. Previously, mostly cross-sectional MRI studies in healthy siblings as well as first-degree relatives have shown inconsistent findings (
Table 1). Similarly, many studies of ultra high-risk individuals have also failed to show consistent hippocampal loss prior to the onset of psychosis (
30,
32,
33,
60). Many of these studies, including those of high-risk populations, have included patients who have some schizophrenia spectrum symptoms or have been exposed to antipsychotic medications, which could have resulted in some of the inconsistencies. In a separate pilot analysis, we attempted to address this issue by comparing hippocampal (total, left, and right) volumes in medication-naive siblings of schizophrenia spectrum patients (N=15 [24 scans]) with volumes in healthy comparison subjects (N=15 [24 scans]). The siblings, which were probably comparable to an ultra high-risk group with schizotypal symptoms, also failed to show hippocampal volume reduction (p=0.7 [unpublished data available upon request from A. Mattai]). Overall, these findings strongly suggest the state-/disease-dependent nature of hippocampal volume loss. Strengths of the present study are the large sibling cohort, which enabled the selection of truly healthy comparison subjects, and the longitudinal nature of the study, which strengthened the stability and significance of the findings.
The effects of antipsychotic medication on hippocampal volume have been addressed by a few studies. The single longitudinal study (N=107) showed unchanged anterior hippocampal volume in patients regardless of cumulative antipsychotic dose (
61). A cross-sectional study (N=56) showed that atypical antipsychotics rather than haloperidol were associated with larger hippocampal volumes after controlling for disease severity (
62). On the other hand, studies of hippocampal volume in antipsychotic-naive and minimally medicated first-episode schizophrenia patients showed that hippocampal volume deficits were present at the onset of schizophrenia prior to any treatment (
9,
63). All of our patients were exposed to antipsychotic medications, but the volume deficits remained fixed throughout the age range, suggesting minimal medication influence at least on the developmental trajectory, which we have also seen in the cortex (
64). Although the effect of medications cannot be definitively ruled out with these observations, combined with the lack of volume loss in siblings, they support evidence that medications have minimal effect on hippocampal volume loss. A direct comparison of medicated and medication-naive childhood-onset schizophrenia patients may address this issue more definitively, but drug-naive childhood-onset schizophrenia patients are almost impossible to recruit.
Postmortem work investigating morphometric hippocampal changes in schizophrenia suggests that the illness reduces hippocampal neuronal size. Benes et al. (
12) measured pyramidal neuron size in the posterior hippocampus and found reductions of 13%–18% in regions CA1–CA4 in schizophrenia patients relative to comparison subjects. Correction for the effects of age, fixation interval, and neuroleptic exposure did not alter the results. Similarly, Arnold (
65) reported reductions in neuronal size in hippocampal subfields that mediate interactions with the cortex, thus possibly altering the neural circuits (
66). Processes occurring during embryonic development and early childhood, such as neuronal migration, neuron enlargement and differentiation, and apoptosis in brain maturation, all have some bearing on hippocampal cytoarchitecture and neuronal arrangement (
65). While it is not clear which of these factors may cause volume loss in the hippocampus, the nature of these neuropathologic changes suggests that at least part of the hippocampal disease process occurs during early development.
Given that the primary development of the brain occurs during fetal life, adverse environmental variables in early life could affect hippocampal development. Across environmental variables, obstetrical complications are one of the strongest predictors of risk for schizophrenia (
67–
70). There is persuasive evidence to suggest that obstetrical complications, particularly perinatal hypoxia and prenatal infections, are related to smaller hippocampal volumes in schizophrenia (
71,
72). Furthermore, in animal models, prenatal- and birth-related hypoxic insults have been demonstrated to result in hippocampal neuron damage and a reduction in hippocampal cell number (
73,
74).
Studies of hippocampal volume in monozygotic and dizygotic twins discordant for schizophrenia have also bolstered support for the idea that hippocampal volumes are differentially modulated by environmental factors to a greater degree when compared with healthy individuals (
75,
76). However, a small retrospective chart study found no evidence for increased obstetrical risk in childhood-onset schizophrenia versus sibling comparison subjects (
77). Collectively, such work highlights that unique environmental events can significantly influence hippocampal volume in patients with schizophrenia.
A model of the developmental pathology of the hippocampus in childhood-onset schizophrenia could consist of an early environmental risk factor, such as perinatal hypoxia or prenatal infections, imparting a constitutional vulnerability to the hippocampus. Studies have shown that the hippocampus is particularly susceptible to damage as a result of stress (
78,
79). During early adolescence, the hippocampus regulates the hypothalamic-pituitary-adrenal axis that releases cortisol and consequently augments dopamine activity in certain brain regions, including the mesolimbic system (
80,
81). Increased stress could lead to an increased demand placed on the hippocampus, eventually leading to an exaggerated response to stress and more hippocampal damage in a positive feedback system. Given the pronounced prefrontal cortical deficits seen in childhood-onset schizophrenia (
38), the prefrontal cortex may have limited ability to take over functions, such as working memory, from the hippocampus, further increasing functional demands and leading to hippocampal damage. This framework could explain a hippocampal diathesis-stress model in which normal maturational processes and exaggerated responses to early stress lead to abnormal hippocampal development that could be static with continued illness burden.
Our findings have several broad-ranging implications in terms of prevention and treatment. First, given the potential significant environmental contribution to hippocampal volume in schizophrenia, measures to decrease exposure to the environmental influence could result in a reduction in the incidence of illness in the population. For example, Suvisaari et al. (
82) found that a decline in bacterial illnesses and initiation of immunization programs may have led to a decline in the incidence of schizophrenia in Finland since the 1950s. In addition, prenatal and perinatal monitoring may also decrease the risk of schizophrenia in some genetically at-risk individuals (
83). As hippocampal pathology likely begins and progresses in early brain development, measures to attenuate or reverse volume loss should be initiated early. The hippocampus is one of few brain structures with the ability to generate new neurons throughout its life (
84). Although there is limited work linking schizophrenia to decreased hippocampal neurogenesis or on whether normalization of neurogenesis would improve atrophy (
85), recent investigations suggest that exercise may promote hippocampal plasticity and improve memory (
86,
87). While many obstacles need to be overcome, future translational studies should focus on early interventions, possibly in the fetal period, as a way to improve hippocampal development and potentially prevent or delay onset of illness.
A major limitation to this study is that we did not investigate hippocampal shape abnormalities that may have reflected on heterogeneous changes within the hippocampus. Shape and subregional analyses of the hippocampus within this population are ongoing. Another limitation of the study is the unknown bias of national recruitment for this very rare patient population that may favor healthy families and thus a population with lower genetic risk. Nonetheless, this study highlights that hippocampal deficits are not strongly related to genetic factors and may represent an important intermediate disease phenotype in childhood-onset schizophrenia.