Converging neurobiological evidence suggests that ADHD involves alterations of catecholaminergic brain circuits (
6). Neuroimaging studies in children with ADHD also show neurodevelopmental brain anomalies, such as reduced cortical volume and folding and gray matter heterotopia (
7–
10), which indicate that, neuroanatomically, ADHD is a disorder of early brain development. In addition, specific neuroimaging modalities, such as diffusion tensor imaging (
11–
13), (resting-state) functional magnetic resonance imaging (fMRI) (
14–
16), and EEG (
17), have shown that both structural and functional brain connectivity are impaired in ADHD patients.
To date, eight independent genome-wide linkage scans have been conducted for ADHD (
23,
24). A recent meta-analysis of seven of these studies identified genome-wide significant linkage for a chromosomal region on 16q (
23). In addition, a large number of candidate gene association studies have been published, and these have primarily focused on genes involved in catecholaminergic neurotransmission. Recent meta-analyses of these studies have yielded significant meta-association for six ADHD candidate genes (i.e., the dopamine and serotonin transporter genes
DAT1/
SLC6A3 and
5-HTT/SLC6A4, the
DRD4 and
DRD5 dopamine receptor genes, the
HTR1B serotonin receptor gene, and the
SNAP25 gene involved in neurotransmission) (
25,
26). In 2008, the results of three genome-wide association studies (GWASs) for ADHD were published (
27–
29) in two independent data sets. Lasky-Su et al. (
28) reported the following two findings that reached (trait-specific) genome-wide significance: 1) a single nucleotide polymorphism (SNP) in
CDH13 and 2) a SNP in
GFOD1.
In the present study, we attempted to integrate the most significant findings from the five reported GWASs for ADHD into protein signaling networks that would not only increase our understanding of the genetic etiology of ADHD but also provide clues for future research into more effective (psychopharmacological) treatments for the disorder. Based on bioinformatics analyses and a systematic literature analysis of the (putative) function of the proteins encoded by the 85 top-ranked genes emerging from the five GWASs for ADHD, we describe a signaling network involved in neurite outgrowth that includes 45 of these proteins. The other 40 proteins could not be convincingly linked to the network. Corroborating evidence for the involvement of this gene network from chromosomal aberrations in humans, from animal models, and from psychopharmacological studies is also presented.
Results
Table 1 shows the 85 genes from the five GWASs for ADHD fulfilling the inclusion criteria of the present study. When more than one SNP in a single gene was found among the top findings, only the SNP yielding the lowest p value for association with (a quantitative phenotype of) ADHD is presented. As can be derived from
Table 1, the only overlap between the top findings of the five published GWASs was for
CDH13, in which (three different) SNPs were associated with ADHD at p<1.00E-04 in three GWASs (
28,
29,
31).
Analysis of these 85 top ADHD candidate genes using the Ingenuity pathway software revealed a significant enrichment (p=4.11E-08 after correction for multiple testing) of the functional gene category neurological disease, with 44 of the 85 genes falling into this category (
Table 2). Furthermore, analysis with the BiNGO bioinformatics tool revealed that the gene ontology processes (calcium) ion binding and hexokinase activity were significantly enriched in the 85 ADHD genes (2.94E-03<p<5.43E-03 and p=2.69E-02, respectively) (
Table 2).
Further literature analysis of the (putative) function of the proteins encoded by the genes in the enriched BiNGO terms revealed that 21 out of 36 genes in the (calcium) ion binding category and two out of two hexokinase activity genes play a role in neurite outgrowth. Subsequently, we investigated the function of the entire set of 85 genes further and found that a total of 45 of the 85 included genes (indicated in bold in tables, figure legend, and throughout this article) fit into a neurodevelopmental network that is involved in directed neurite outgrowth, which is illustrated in
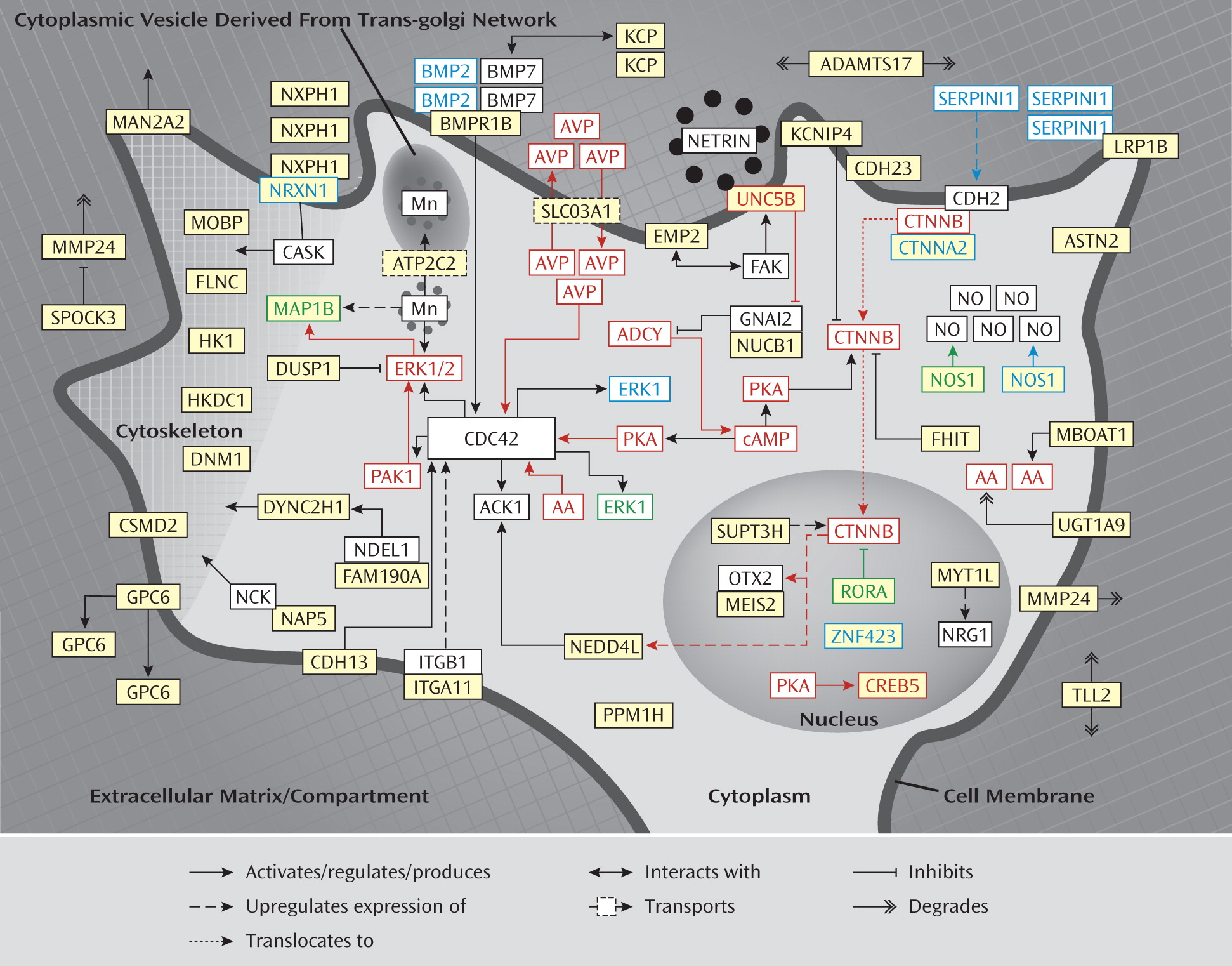
Figure 1. The proposed signaling network links axonal guidance and neuronal cell adhesion proteins at the neuronal cell membrane with downstream acting adaptor proteins and neuronal cytoskeleton/extracellular matrix-associated proteins.
A detailed description of the evidence linking the genes in the network to neurite outgrowth is provided in the data supplement accompanying the online version of this article.
Involvement of the identified network in ADHD is also supported by the finding that several of the identified genes (i.e.,
CTNNA2,
NRXN1,
NAP5,
SERPINI1,
NOS1,
ERK1,
ZNF423,
NEDD4L, and
BMP2) are located within copy number variations-and are hence deleted or duplicated-in people with ADHD (
20–
22,
35–
41) (
Table 3,
Figure 1).
Moreover, partial knockout models of the
NOS1,
ERK1,
MAP1B, and
RORA genes in mice provide further evidence of the involvement of these genes in ADHD etiology (
Figure 1).
Mice in which the
Nos1 and
Erk1 genes have been knocked out display hyperactive behavior (
42,
43), while, in contrast,
Map1b- (
44) and
Rora-knockout mice (
45) show hypoactivity.
Lastly, several proteins from the network appear to be under control of the stimulants methylphenidate and amphetamine that are used to treat ADHD symptoms (
4,
5). Both stimulants have been shown to stimulate neurite outgrowth (
46,
47) and directly or indirectly regulate the expression and/or function of several genes/proteins implicated in neurite outgrowth, including a number of genes/proteins from the identified network (
Figure 1).
Methylphenidate exposure upregulates the expression of
AVP (
48) and
CREB5 (
49) (
Figure 1). In addition, methylphenidate upregulates the expression of a large number of other genes not directly found in the identified network but functionally implicated in neurite outgrowth (
50). Examples of these genes are
MMP14,
TIMP2, and
TIMP3. The
MMP14 gene encodes a neuronal extracellular matrix metalloproteinase, such as
MMP24, and
TIMP2 and
TIMP3 encode two direct inhibitors of MMP14 (
34,
50). Amphetamine upregulates the expression of
UNC5B (
51) and
PAK1 (
52) and downregulates the expression of
CTNNB (
53). Amphetamine also activates neuronal ERK1 and ERK2 (
54) and increases the level of arachidonic acid in neocortical, extrapyramidal, and limbic brain regions (
55). Deficiencies or imbalances of arachidonic acid have been shown to be associated with ADHD (
56). Lastly, the adenylate cyclase/cAMP/protein kinase A/CREB-signaling cascade (including protein kinase A activating
CREB5) is also activated by amphetamine (
57).
An important potential bias of which to be aware in the analysis of the top findings from the reported GWASs of ADHD is the fact that large genes, which are often brain expressed, may be more likely found as associated with a phenotype in a GWAS as a result of chance, since more SNPs are present in these large genes (
58). Indeed, the genes found among the top findings of the ADHD GWASs were considerably larger than the average gene size of the human genome ([
Table 1] average size of 85 ADHD genes: 373 kb versus average gene size in human genome: 27 kb [
59]).
If large genes are more likely found to be associated with a phenotype of a GWAS because of chance, this would be expected to be the case for GWASs of both nonpsychiatric disorders and ADHD, and therefore we compared the results of our analyses with the Ingenuity and BiNGO software tool results for the 20 top ADHD candidate genes with those for the top 20 candidate genes from four published GWASs for diabetes mellitus type I and Crohn's disease (
Table 4,
Table 5 [also see the data supplement]). This comparison showed that the neurological disease category was not enriched in Crohn's disease and only contained a single gene in diabetes type I (
Table 4). In addition, the gene ontology processes that were enriched in the top GWAS findings were clearly different for the three diseases (
Table 5).
Discussion
In the present study, we used both bioinformatics tools and manual literature mining to attempt to integrate the top findings of the currently available GWAS data for ADHD. We found that 45 of the 85 top ADHD candidate genes (or approximately 53%) fit into a network that is involved in neurite outgrowth. It should be noted that our decision to only include SNPs within exonic or intronic regions of genes and those with association at p<1.00E-04 is essentially arbitrary. By only including genes with SNPs in exons and introns, we have ignored the possible role of upstream and downstream regulatory sequences (
61), which could have increased the number of (potentially relevant) genes to be included in the study. However, we do not expect that this would have altered the general tendency in our results (i.e., the considerable overrepresentation of neurite outgrowth-related genes).
Surprisingly, the enrichment of neurite outgrowth-related genes was not reflected by the finding of gene functional categories and/or gene ontology terms directly related to neurite outgrowth with the bioinformatics tools used in this study (
Table 2). This shows the limitations of the currently available bioinformatics software, which is a result of the current incompleteness of the annotation of the human genome.
Therefore, use of bioinformatics tools should always be accompanied by manual and systematic literature and database mining in order to fully understand the processes involved in the etiology of multifactorial disorders. Moreover, the enrichment of neurite outgrowth-related genes in the ADHD GWAS data (53%) should be viewed from the perspective that only 3% (N=576) of all currently annotated human genes (N=18,589) are involved in neurogenesis (gene ontology term: GO:0022008).
Several additional lines of evidence suggest that the identified network provides an important contribution to the etiology of ADHD. Apart from implicating the same genes as the GWASs, these other lines of evidence point to involvement of several other genes (and their corresponding proteins) that were not found (at the p<1.00E-04 level) in the GWASs but that functionally connect the genes from the GWAS findings.
First, nine proteins that act in the identified neurite outgrowth network are encoded by genes that were also found in copy number variations in people with ADHD (
20–
22,
35–
41) (
Table 3). Second, knockout mouse models of four network genes reveal ADHD-related behavior (
42–
45). Third, and most importantly, the expression and/or function of a number of genes/proteins in the identified network is regulated by the stimulants methylphenidate (
48–
50) and amphetamine (
51–
55,
57), which are the most commonly used psychopharmacological treatments for ADHD symptoms (
4,
5) and directly regulate neurite outgrowth (
46,
47). The latter finding might not only increase our understanding of the working mechanism of these drugs but also provide directions for future research into new and more effective psychopharmacological ADHD treatments.
The fact that stimulants regulate neurite outgrowth potentially implicates a number of classic ADHD candidate genes in our network. Methylphenidate and amphetamine mediate effects on both dopaminergic and serotonergic neurotransmission in the brain (
4,
5,
62). In this respect, the genes encoding the SLC6A3, DRD4, DRD5, SLC6A4, and HTR1B proteins could be putatively linked to the identified network. In addition, the link of these classic candidate genes with the neurite outgrowth network may help to localize the network to dopaminergic and serotonergic brain regions.
The findings from this study fit very well with literature about disturbed brain structure and function in ADHD patients (
7–
10) and, even more so, with recent reports describing aberrant structural and functional brain connectivity in these patients (
11–
17). Because differences in brain connectivity have been shown to underlie interindividual variability in complex cognition-related processes, including ADHD (
63,
64), relatively minor alterations in neurite outgrowth efficiency or direction may provide a major contribution to the cognitive deficits observed in ADHD.
A possibly important bias in our study may arise from the fact that the brain-expressed genes in the top findings of the ADHD GWASs are substantially larger than the average gene size in the human genome, and seven of the nine very large genes (i.e., genes with a size of >1 Mb) from the list of 85 ADHD genes indeed encode proteins that fit into the proposed ADHD network. It has been argued that the overrepresentation of large genes in the top findings of GWASs might result from bias because one is more likely to find an associated SNP in a larger, usually brain-expressed gene (
58). If this is the case, it would be expected that similar results would be found in GWASs of polygenic disorders that are assumed not to primarily originate in the brain, such as diabetes mellitus and Crohn's disease. However, our Ingenuity and BiNGO analyses on the 20 top candidate genes from the GWASs for ADHD, diabetes mellitus type I, and Crohn's disease (
Table 4,
Table 5) show that this bias does not likely explain the findings for ADHD. In this respect, we would like to submit that the strong enrichment of neurite outgrowth-related genes in the top findings from the five ADHD GWASs, with 45 out of 85 genes fitting into our proposed network, also argues against the fact that these genes are spurious association findings. Nevertheless, we cannot completely exclude the possibility that some of the genes placed in the network still might have been chance findings.
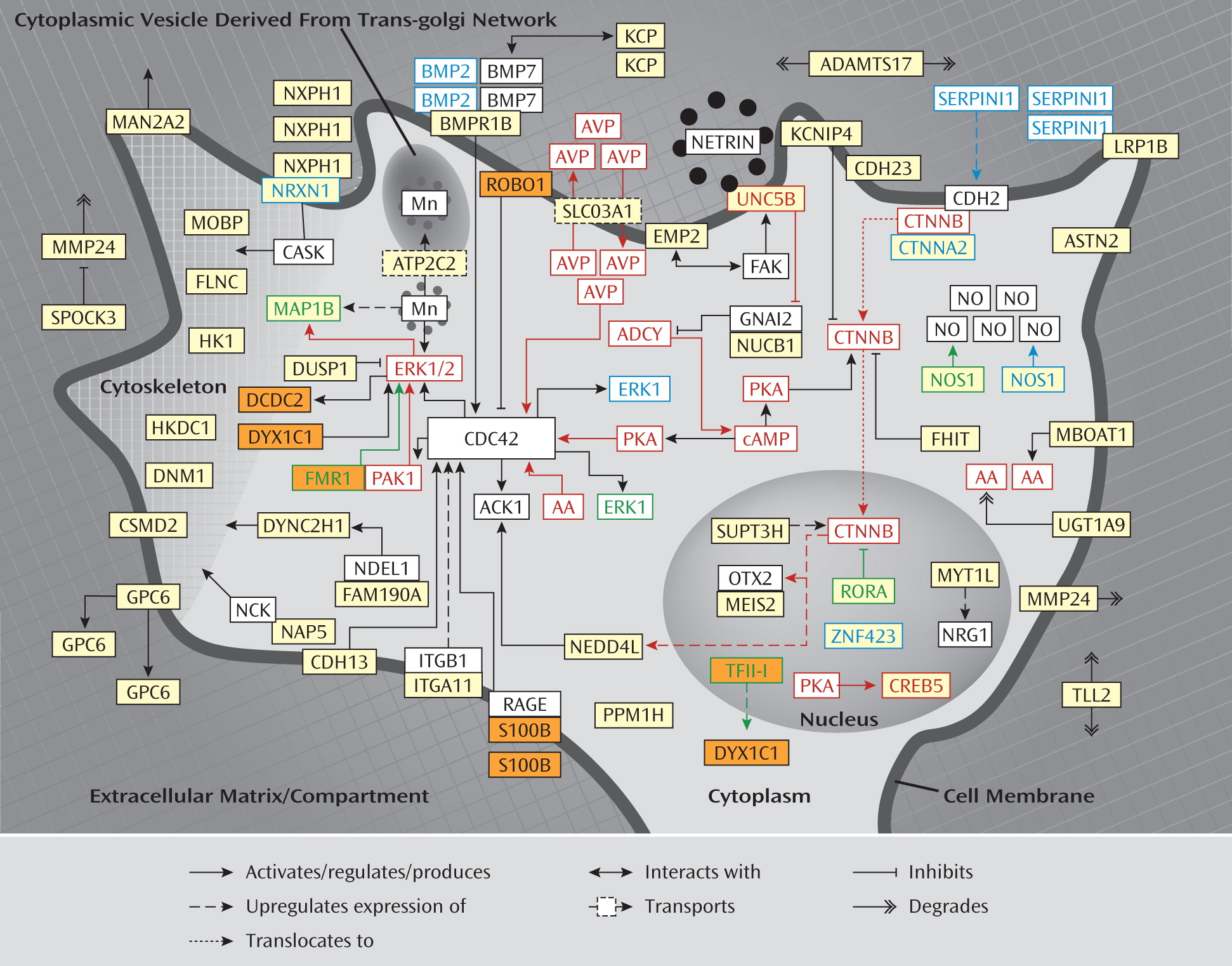
Is the involvement of neurite outgrowth genes specific to the etiology of ADHD? This seems rather unlikely, since we recently also found neurite outgrowth to be implicated in dyslexia (
65). Following an approach similar to the one used in the present study, we found that 10 of 14 dyslexia candidate genes reported to date (i.e.,
DCDC2, DIP2A, DOCK4, DYX1C1, FMR1, GTF2I, KIAA0319, KIAA0319L, ROBO1, S100B ) fit into a protein network that is involved in neurite outgrowth and neuronal migration. As shown in
Figure 2 and explained in further detail in the data supplement, the proteins encoded by six of these genes (i.e., DCDC2, DYX1C1, FMR1, TFII-I [encoded by
GTF2I], ROBO1, and S100B) fit directly into the identified network for ADHD. It is also interesting to note that decreased serum concentrations of S100B correlate with increasing hyperactivity symptoms in children with ADHD relative to normal comparison subjects (
66) and that knockout mouse models of the
FMR1 (
67) and
GTF2I (
68) genes show ADHD-like behavior. One possible explanation for the observed genetic overlap between ADHD and dyslexia is that these neurodevelopmental disorders are highly comorbid and genetically correlated with one another (
69–
71). The same also holds true for autism (
72), and recent evidence suggests involvement of neurite outgrowth genes in this neurodevelopmental disorder as well (
73). The different functional consequences of disturbed or abnormal neurite outgrowth in the different brain regions that are most affected in ADHD, dyslexia, and autism, respectively, could help to explain why a disturbance of the same neurodevelopmental process could lead to these partially overlapping but still distinct clinical phenotypes.
Several genes that are major players in the identified neurite outgrowth network have not been directly observed in the top findings of the GWASs. An example of such a gene is
ERK1, which not only has an important function in the network but has also been implicated in ADHD through its location in ADHD-related copy number variations (22, 38) as well as the fact that
Erk1-knockout mice display hyperactive behavior (
43) and that amphetamine directly activates neuronal ERK1 (
54). Other examples are
CTNNB and
CDC42. These and other genes from the network may be strong candidates for future association studies.
Lastly, our data make a compelling case for the use of polygenic types of association analyses to explain a higher percentage of the heritability of ADHD through the GWAS findings. In this respect, all the individually associated genes from the identified neurite outgrowth network could be fitted simultaneously into one polygenic risk test, as illustrated in the recent study by Yang et al. (
74) of all SNPs that have been individually associated in GWASs of variation in human height as well as in two other recent studies applying similar approaches to GWAS data for schizophrenia (
75) and functional gene groups for cognitive ability in ADHD (
76).
In conclusion, by integrating the top findings of the five GWASs for ADHD with copy number variation data, (psychopharmacologically induced) expression data, and data from animal studies, we have identified a protein signaling network for ADHD that results in directed neurite outgrowth. Systems biology approaches like those used in this study are needed to yield genetic findings that are useful for clinical purposes, such as the prediction of disease prognosis and the identification of new treatment targets and strategies.