Major depressive disorder is a debilitating disorder of low affect and altered mood regulation that affects approximately 17% of the population at some point in life, resulting in serious personal, social, and economic burdens (
1). The prevalence of major depressive disorder is two times higher in women than in men. Female patients with the disorder tend to have higher symptom numbers, a more severe type of depression, and greater risk of recurring episodes compared with male patients, but the underlying biological vulnerabilities have not been characterized (
2). Changes in the structure, function, and coordinated activity of several brain regions may underlie impaired mood regulation in depression (
3). Increased metabolic activity in one of these regions, the subgenual anterior cingulate cortex, has been consistently reported in the induction of the depressive state, and subgenual anterior cingulate cortex metabolism is reversed by pharmacological treatment (
4) and deep brain stimulation (
5).
Low neurotrophic support in limbic brain regions has been proposed as a unifying hypothesis for the reduced density or cell numbers in the frontal cortex (
6) and amygdala (
7) and the reduced hippocampal volume observed in individuals with major depression (
8). Rodent studies have demonstrated that various antidepressant treatments increase brain-derived neurotrophic factor (
BDNF) expression (
9), and BDNF infusion into the hippocampus is sufficient to produce an antidepressant-like effect (
10). Despite abundant animal studies supporting the close relationship between BDNF and depression, direct evidence in humans is limited to reports of low circulating peripheral BDNF levels, which are normalized by antidepressant treatment (
11), and studies demonstrating reduced pro-BDNF and BDNF levels in the postmortem amygdala of depressed female subjects (
12) and in the hippocampal tissue in depressed patients (
13,
14). Additionally, studies have reported that individuals who die by suicide exhibit low hippocampal and midbrain BDNF levels (
15), reduced activity-dependent
BDNF expression by hypermethylation of promoter/exon IV of the
BDNF gene (
16), and, in carriers of the
BDNF Met allele, increased risk for violent suicide (
17,
18), together providing additional evidence that BDNF has a role in the psychopathology of major depression.
In parallel, human imaging and basic science studies have suggested excitation/inhibition impairment in individuals with major depression that is potentially mediated by decreased GABA content (
19). We recently reported down-regulation of several GABA-related genes in the dorsolateral prefrontal cortex (
20), subgenual anterior cingulate cortex (
21), and amygdala (
12) in patients with major depression, potentially affecting somatostatin-positive dendritic targeting interneurons. We further demonstrated that a set of amygdala-related gene changes (affecting the
TAC1,
CORT,
NPY,
SST,
RGS4, and
SNAP25 genes [
Table 1]) correlate with reduced BDNF expression in depressed patients and in mice with reduced BDNF function, hence identifying a pattern of reduced BDNF-dependent gene expression in major depression (
12) and providing supporting evidence for a link between the neurotrophic (
9) and GABA (
19) hypotheses implicated in depression.
Discussion
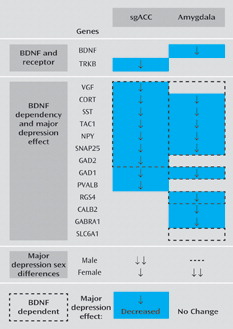
Seeking molecular evidence in support of a low BDNF and reduced GABA function pathway in the subgenual anterior cingulate cortex in individuals with major depression, we first relied on two strains of mice with reduced BDNF function to determine the extent to which our genes of interest depend on BDNF for expression in the cingulate cortex (
Table 1). Translating this information to human subjects with major depression and measuring expression levels in their subgenual anterior cingulate cortex, we observed no changes in
BDNF itself, reduced expression of
TRKB, the main receptor through which BDNF signals, and reduced mRNA levels of several genes for which expression depends on BDNF (
Table 2). Among these BDNF-dependent and depression-affected genes, the reduced expression of several markers of GABA-ergic interneurons that specifically target the dendritic compartment of pyramidal neurons (
SST, NPY, and
CORT) suggests the presence of a reduced dendritic inhibition phenotype in individuals with major depression, downstream from low BDNF signaling. GABA-related changes also extended to genes with modest or no evidence for BDNF dependency (based on mouse studies [also see
Table 1 and
Table 2]), suggesting that additional factors lead to reduced GABA function in depression. Overall, results were more robust in men, which is contrary to our previous observations in the amygdala, in which depressed women exhibited reduced
BDNF levels (but not
TRKB levels) and greater BDNF-dependent gene changes compared with men. Together, these results suggest a core BDNF-/GABA-related pathology in major depression that affects markers of interneurons targeting pyramidal cell dendrites and that displays sex- and brain region-specific features.
With the exception of sex and, to a lesser extent, age, none of the clinical, demographic, and technical parameters had any consistent detectable effects on gene expression in our relatively large cohort of human subjects. Death by suicide has been associated with reduced BDNF expression, but differences observed in the present study appeared to be more robust in subjects who did not die by suicide, compared with the respective comparison subjects (see Table S3 in the online data supplement).
When the groups were segregated by sex, we observed similar changes in
TRKB expression and overall lower statistical significance of changes in depression-related gene down-regulation in female subjects, although not systematically (
Table 2). Notably, the expression levels of three BDNF-dependent genes (
CORT, NPY, and
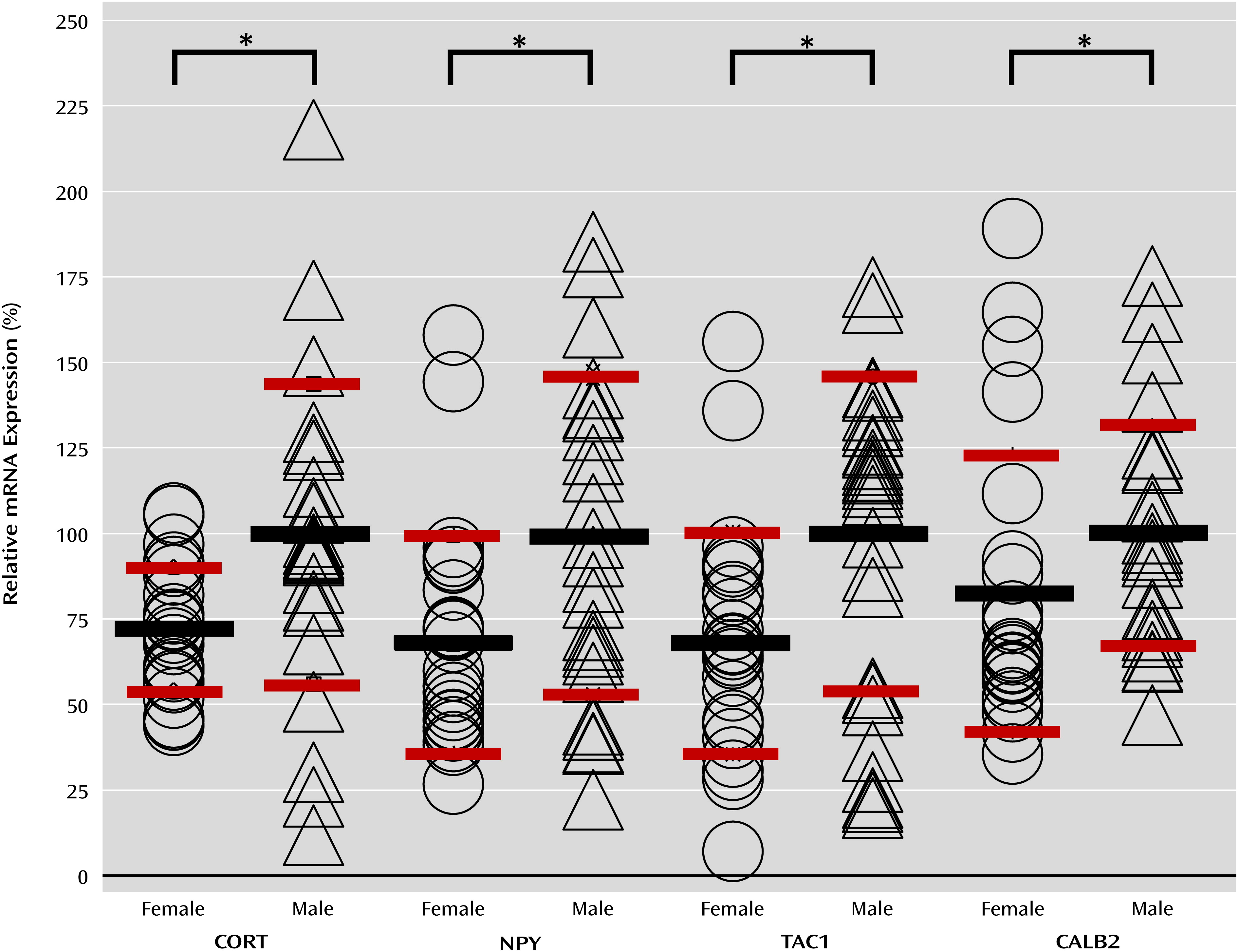
TAC1) were already lower in female comparison subjects relative to male comparison subjects (
Figure 1). Despite lower baseline levels, expression changes for
CORT,
TAC1, and
NPY still displayed greater or equal statistical significance and effect size in depressed female subjects compared with depressed male subjects. Thus, despite a less robust profile of molecular changes, the low female baseline expression for some genes may result in a greater propensity to reach the threshold of low pathophysiological function (
Table 2). Finally, the overall male-female similarities in gene changes downstream from low BDNF in the two mouse strains (
Table 1) and the reduction or absence of changes in human female subjects for genes with intermediate and low BDNF dependency (
SNAP25,
GAD1/
GAD2 [
Table 2]) suggest the presence of additional sex-specific moderating factors in human subjects. This is consistent with our previous study of the amygdala in depressed male (
23) and female (
12) subjects, in which BDNF and BDNF-dependent genes were robustly affected in women but not in men. Together, these results support the concept that sex differences in the vulnerability to and the expression of major depression may not result from different pathophysiological mechanisms but rather from moderating biological factors acting on a core phenotype implicating a putative reduction in GABA function and dendritic targeting interneuron vulnerability.
SST,
NPY, and
CORT are three neuropeptide coding genes with overlapping patterns of expression in mice that are found in approximately 20% of interneurons and that have the functional characteristic of providing GABA-mediated inhibition to distal dendrites of pyramidal neurons. Moreover,
TAC1, the fourth neuropeptide coding gene that is similarly down-regulated in the subgenual anterior cingulate cortex and amygdala in individuals with major depression (
Figure 2), encodes for substance P, a gene product with putative antidepressant activity (
27), which in the cortex mainly activates SST-positive cells through NK1R receptor binding (
28). Since all four genes are dependent on BDNF for their expression (
Table 1), low BDNF signaling may orchestrate a synergy between decreased
TAC1, SST, NPY, and
CORT expression, leading to reduced inhibition onto the dendritic trees of targeted pyramidal neurons.
VGF and
SNAP25, two genes involved in synaptic function and previously implicated in major mental illnesses, were also found to be BDNF dependent and down-regulated in the subgenual anterior cingulate cortex in subjects with major depression, suggesting that a broader BDNF-dependent module may be affected. Finally, expression of
GAD1, a gene encoding an enzyme that produces GABA, and expression of
PV, a gene encoding a marker for fast spiking GABA interneurons targeting the cell body and axon initial segment, were also down-regulated. For
PV and
GAD1, the mechanism appears to be independent of reduced BDNF function (
Table 1,
Table 2). Notably,
PV levels were not affected in other brain regions (the dorsolateral prefrontal cortex [e.g.,
20] and the amygdala [e.g.,
12]), and calretinin (
CALB2), a marker for a third interneuron subset, displayed reduced baseline expression and no depression-related changes in the subgenual anterior cingulate cortex but was decreased in the amygdala in depressed female subjects (
Figure 2). Together, these findings put forward critical observations of the pathology of major depression, which may relate to three consecutive biological scales: 1) molecular function, manifested by altered BDNF-/TrkB- and GABA-associated gene function; 2) cellular microcircuitry, in which findings appear to be clustered by function (i.e., dendritic inhibition); and 3) circuit moderators, in which sex-related factors and brain regions are relevant modulators to gene expression in major depression. Hence, reduced GABA-mediated inhibition of incoming information in pyramidal dendrites may represent a putative microcircuitry-level phenotype underlying the increased activation of the subgenual anterior cingulate cortex and amygdala that is frequently reported in studies of patients with major depression (
29,
30). In turn, restoring dendritic inhibitory function may reduce pyramidal cell activation and excitatory tone and contribute to the reduction in activation of the subgenual anterior cingulate cortex with positive treatment response to therapeutic intervention (e.g., deep brain stimulation, antidepressants) (
4,
5).
Some of the limitations of these results are inherent to investigation of heterogeneous cohorts and of postmortem brain samples. Large numbers of clinical, demographic, and technical parameters have to be taken into consideration, and results are mostly correlative and cannot provide insight into developmental processes in major depression. Our relatively large cohort size allowed us to rule out major effects of putative confounds (details are summarized in Table S2 in the online data supplement), but the results will need to be confirmed in independent cohorts.
The causal link between reduced BDNF signaling and altered gene expression was inferred from analyses of rodents with genetically induced reduction in BDNF function, but species differences may exist, and thus the different labels of gene-specific BDNF dependency may vary. The fact that regional variations in BDNF dependency were also observed in rodents suggests that aspects of the human gene regulation patterns are conserved across species. This latter observation supports the need for further studies of rodent models with more refined genetic manipulations affecting specific interneuron populations, for instance, to assess the effect on reduced dendritic inhibition on the local microcircuitry in the amygdala and cingulate cortex and downstream behavioral phenotypes. Indeed, it is evident that the complexity of the putative BDNF-mediated cellular and signaling phenotype observed in human major depression is not fully replicated in currently available genetic rodent models, and thus caution should be applied when interpreting rodent behavioral outputs downstream from broad genetic changes; for instance,
Bdnf+/− and
BdnfKIV mice exhibit normal and increased emotionality, respectively (
31,
32). Disruption of forebrain-specific BDNF leads to higher emotionality when combined with exposure to chronic stress in female mice (
33,
34). Conversely, low ventral striatum BDNF can have antidepressant-like effects (
35). Our observation in humans of reduced expression of
BDNF in the amygdala and of reduced
TRKB in the subgenual anterior cingulate cortex suggests that altered BDNF signaling may represent a complex integration of currently unidentified upstream events (e.g., stress factors, developmental trajectories, and genetic variation), which result in similar core downstream changes (i.e., reduced markers of dendritic inhibition) and that are further moderated by numerous factors (e.g., sex, brain region, and brain activity).
Finally, it is also becoming evident that the observed findings are not specific to major depression, since similar reductions in
BDNF and
SST expression have been reported in studies of other neuropsychiatric (e.g., schizophrenia [
36,
37] and bipolar disorder [
38]) and neurological (e.g., Alzheimer’s disease and Huntington’s disease) disorders. Thus, our findings may more accurately reflect a molecular and cellular endophenotype that implicates BDNF signaling and GABA microcircuitry and that has its own etiological factors. However, the restricted scope on markers affecting dendritic inhibition that we observed differs from observations in studies of other diseases in which changes occur in the context of other core pathologies, such as robust
PV-related GABA dysfunction in schizophrenia (
39) or neurodegenerative processes in Alzheimer’s disease (
40). Investigating the etiological factors and phenotypic outputs of these respective molecular and cellular endophenotypes outside the restriction of the categorical definitions of psychiatric and neurological illnesses may provide dimensional insight into relevant proximal pathophysiological mechanisms to be targeted for therapeutic purposes, while their patterns of co-occurrences may be informative of mechanisms underlying clusters of symptoms that are enriched in clinically defined disorders.