Several lines of evidence implicate white matter abnormalities in the pathophysiology of bipolar disorder. Postmortem studies have shown reduced numbers of oligodendrocytes (
1) and compromised myelin (
2). Additionally, decreased expression of myelin and glia-related genes have been reported in patients (
3). Using nondiffusion MRI, reduced white matter volume and density have been observed for global and regional measures, particularly in the corpus callosum, internal capsule, and temporal lobes (
4). However, these methods are suboptimal for identifying white matter pathology. In contrast, diffusion tensor imaging quantifies fractional anisotropy, which, although nonspecific to any particular underlying cellular pathology, is a sensitive marker of general white matter integrity as observed in model organisms (
5) and humans (
6). While reduced fractional anisotropy is repeatedly reported in bipolar disorder (
4,
7), the precise locations of these deficits tend not to replicate across studies, possibly reflecting limited sample sizes and clinical heterogeneity. Moreover, the nature of white matter pathology in bipolar disorder is unknown. Anisotropy reductions could result from genetic liability for the disorder or be associated with manifestation of the illness or its treatment. The present study was designed to disentangle these potential explanations by investigating anisotropy in a large sample of individuals with bipolar disorder, their unaffected siblings, and unrelated comparison subjects.
As traits that lie intermediate between the action of risk genes and clinical diagnosis, endophenotypes enhance our understanding of biological mechanisms underlying disease. To qualify as an endophenotype, a measure must be heritable, altered in the clinical population, state independent, and altered in unaffected relatives (
8). White matter integrity, as indexed by fractional anisotropy, is heritable (
9,
10), and diffusion tensor imaging studies have revealed suggestive evidence of white matter integrity reductions in unaffected relatives (
11,
12). Versace et al. (
13) demonstrated age-by-group interactions in the unaffected teenage offspring of bipolar probands. Mahon et al. (
14) reported reduced fractional anisotropy in the right temporal lobe of 15 unaffected relatives. In a larger sample, Sprooten et al. (
15) found widespread reductions in the relatives of bipolar probands. However, this sample did not include a patient group for direct comparison, and unaffected relatives were young, leaving the possibility that individuals yet to develop the illness drove the effects.
As etiologically informed measures, endophenotypes often highlight sources of clinical heterogeneity within diagnostic categories. A potentially important source of clinical heterogeneity in bipolar disorder is the presence of psychotic symptoms, as implied by differences between psychotic and nonpsychotic patients in D
2 dopamine receptor density (
16), neuroanatomy (
17), functional connectivity (
18), and neurocognitive functioning (
19). These differences could point to unique genetic components that are specific to psychotic bipolar disorder, and they may have implications for psychiatric nosology and treatment efficacy.
In the present study, we examined fractional anisotropy in individuals with bipolar disorder and their unaffected siblings who were past the typical onset age of the illness. Reductions in siblings and intraclass correlations between sibling pairs were interpreted as familial contributions to reductions in the patients, whereas correlations with duration of illness and symptom severity indicated illness-specific mechanisms. Furthermore, associations of white matter integrity with history of psychotic symptoms were investigated to inform the homogeneity of fractional anisotropy against a clinically heterogeneous background.
Method
Participants
Participants provided informed consent and the study was approved by the institutional review boards at Hartford Hospital and Yale University. Overall, 64 remitted patients with bipolar I disorder, 60 full siblings without bipolar disorder, and 46 demographically matched healthy individuals participated (
Table 1). Patients were identified through outpatient clinics and community mental health facilities in the Hartford area. Inclusion criteria for patients were age between 18 and 70 years, diagnosis of bipolar I disorder as determined by the Structured Clinical Interview for DSM-IV (SCID) (
20), and at least one full sibling without bipolar disorder willing to participate in the study. Siblings were included if they were within 10 years of the age of the bipolar proband and did not have a diagnosis of bipolar spectrum disorder. To increase ecological validity of the sample, sibling pairs were not excluded for anxiety disorders, a single episode of major depression, or past substance abuse or dependence. Unrelated healthy comparison subjects were included if they had no lifetime history of axis I psychiatric disorder as assessed by the SCID and no family history of mood or psychotic disorders. Participants were excluded for alcohol or drug abuse or dependence within the past 6 months, a history of major medical or neurological disorders, or IQ <70 as assessed by the WAIS (
21). In patients, euthymia was established with the Hamilton Depression Rating Scale (HAM-D) (
22), the Young Mania Rating Scale (
23), the Brief Psychiatric Rating Scale (BPRS) (
24), and through diagnostic case reviews. Severity of lifetime history of psychosis was assessed with the Lifetime Dimensions of Psychosis Scale (
25), a carefully validated dimensional system for rating longitudinal features of affective and psychotic symptoms.
Scan Acquisition
Scans were collected at the Olin Neuropsychiatry Research Center Institute of Living using a Siemens Allegra 3-T scanner. Diffusion-weighted MR images were acquired using single-shot echo planar imaging (TR=6,300 ms, TE=81 ms, field of view=22×22 cm, matrix=128×128, and voxel size=1.7×1.7×3.0 mm) with a twice-refocusing spin echo sequence to minimize eddy-current induced distortion. Diffusion-weighted volumes were acquired in 55 noncollinear gradient directions at two diffusion weighting values (b=0 and b=800 seconds/mm2), preceded by three non-diffusion-weighted (b=0) images. Each acquisition lasted 6.2 minutes and contained 58 volumes, with 45 contiguous interleaved axial slices per volume (slice thickness, 3 mm) covering the whole brain.
Image Preprocessing
Data were converted to NIFTI format using MRIcron and preprocessed using standard FMRIB’s Software Library tools (
http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/FDT). Images were corrected for subject motion and eddy currents by aligning the diffusion-weighted volumes to each subject’s first b
0-volume. FMRIB’s Software Library brain extraction tool was applied to remove nonbrain tissue. Next, diffusion eigenvectors, eigenvalues, and fractional anisotropy were calculated. The resulting maps were visually inspected. Three scans (one bipolar patient, one sibling, and one comparison subject) were excluded because of signal dropout, and four individuals (two bipolar patients and two comparison subjects) were excluded for hyperintensities.
Tract-Based Spatial Statistics
The 170 fractional anisotropy maps were processed according to the standard pipeline (
26). Briefly, images were eroded and linearly and nonlinearly registered to standard space. Fractional anisotropy images were binarized (>0) and combined to create a study-specific mask containing only voxels present in every image. Maps were masked, averaged, and “thinned,” and the threshold was set at >0.2 to create a study-specific skeleton template. In each individual, the maximum voxel nearby each voxel in the skeleton template was projected onto this template using a distance map, which ensures that the same voxel is only projected onto the skeleton once and that the likelihood of a voxel being projected reduces with increasing distance from the template (
26). This results in one skeleton per subject containing the centers of each individual’s white matter.
Voxel-wise tests between patients and comparison subjects and between unaffected siblings and comparison subjects were performed using threshold-free cluster enhancement (FMRIB Software Library-Randomize) (
27). We corrected p values for multiple comparisons according to family-wise error rate by 5,000 permutations across groups. For all voxel-wise comparisons, age, age squared, and sex were included as covariates, and a significance threshold of p<0.05, family-wise error corrected, was used. Only clusters containing more than 10 contiguous significant voxels are reported. Significant voxels were localized to anatomical structures using standard atlases (
28,
29), followed by an automatic atlas query tool, “autoaq”;
http://brainder.org/2012/07/30/automatic-atlas-queries-in-fsl/). As a cross-validation, voxel-wise statistics were also evaluated using traditional “cluster-extent” statistics, with a cluster-forming threshold of t>2.
Our sample contained 108 full siblings (54 pairs), and intraclass correlations were calculated for the mean values within significant patient < comparison subject and sibling < comparison subject clusters as an index of familiality.
Region-of-Interest Analysis
To extend our voxel-level analysis, atlas-derived regions of interest were examined in three broad categories:
1.
Limbic and frontotemporal connections: column and body of the fornix, cingulum bundles, the hippocampal end of the cingulum, the uncinate fasciculus, and the superior longitudinal fasciculus;
2.
Thalamic tracts: the anterior, posterior and retrolenticular limbs of the internal capsule, the external capsule, and the posterior thalamic radiations; and
3.
Callosal regions: forceps major and forceps minor.
See the
data supplement that accompanies the online edition of this article for detailed methods.
For each region of interest, differences between patients and comparison subjects and between siblings and comparison subjects were tested in separate linear mixed models, with hemisphere as the within-subject factor and group, age, age squared, and sex as covariates. For the callosal tracts, mixed models were calculated with subregion as the within-subject factor. For the fornix, general linear models were calculated with group, age, age squared, and sex as independent variables. We corrected p values for multiple testing according to false discovery rate.
Intraclass correlation coefficients between the sibling pairs were calculated separately for mean fractional anisotropy within each region. Statistics were calculated in R, version 2.15.1 (
http://www.r-project.org/).
Correlations With Clinical Measures and Potential Confounds
Within the patient group, Spearman’s rank correlations were calculated between duration since first manic episode and mean fractional anisotropy across the skeleton and within significant clusters. We also examined the extent to which the effects of duration of illness, duration of illness squared, and age were mutually independent by including age, sex, and age squared in the regression.
Severity of history of psychotic symptoms was estimated by summing each patient’s score for the positive symptoms and schizophrenia subscales of the Lifetime Dimensions of Psychosis Scale. The square root of this composite score (to correct for positive skew in the distribution) was entered in general linear models on mean fractional anisotropy across the skeleton and within significant clusters.
Medication effects were examined by comparing extracted anisotropy values between subgroups of patients who were currently using different classes of medications: antipsychotics, lithium, other mood stabilizers, and antidepressants.
Effects of current nicotine use, anxiety disorders, and past history of alcohol and substance abuse were examined using general linear models with the extracted fractional anisotropy values as dependent variables.
Results
Diagnostic Effects
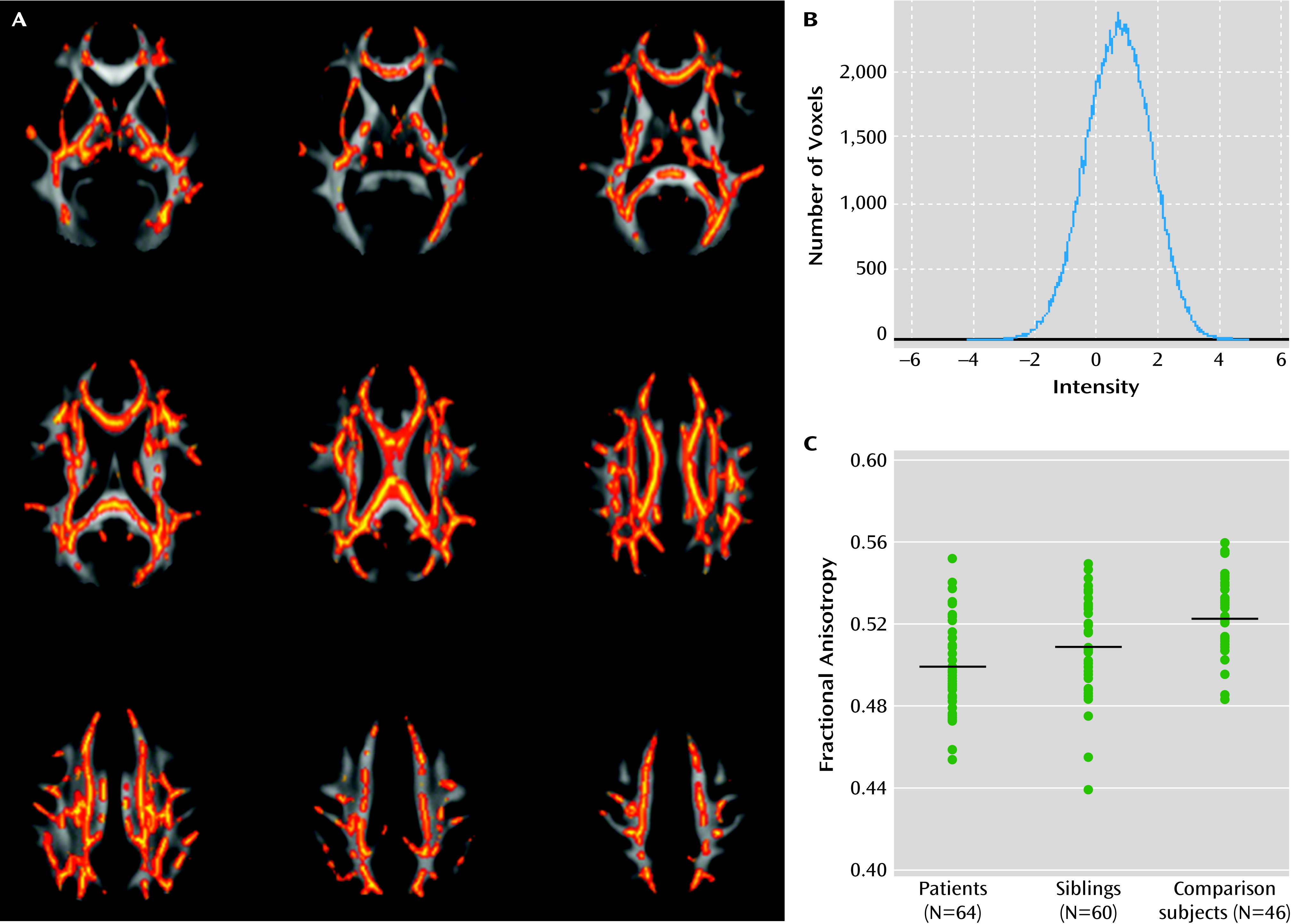
Voxel-wise comparisons indicated that individuals with bipolar disorder exhibited reduced fractional anisotropy across the skeleton in seven clusters (
Figure 1A and see Table S1 in the online
data supplement; p<0.05, family-wise error corrected, df=105). We found no voxels in which fractional anisotropy was increased in patients relative to healthy comparison subjects. T statistics within clusters were between 0.83 and 4.89, and the histogram of voxels in the skeleton was symmetrically distributed with a peak at t=0.74 (
Figure 1B), suggesting that the effect was subtle but spatially consistent. In line with this observation, average fractional anisotropy extracted across the entire skeleton mask was significantly reduced in patients (t=3.26, df=105, p<0.002). Using the more conventional cluster-extent method, we found three smaller clusters restricted to the voxels with the largest effect sizes in the corpus callosum and superior parietal regions (see Figure S2 in the online
data supplement; p<0.05 family-wise error corrected, df=105).
Similar results were obtained when only right-handed individuals were examined, when excluding patients with Young Mania Rating Scale or HAM-D score >7 (N=14), and when we did not covary for age, age squared, and sex.
Region-of-interest analyses also revealed that bipolar patients had significantly reduced anisotropy in most regions (p<0.05, false discovery rate corrected), including the cingulum, superior longitudinal fasciculi, and all thalamocortical tracts except the retrolenticular limb of the internal capsule (
Table 2). No significant group-by-hemisphere interactions were found.
Familiality of Fractional Anisotropy Reductions in Bipolar Disorder
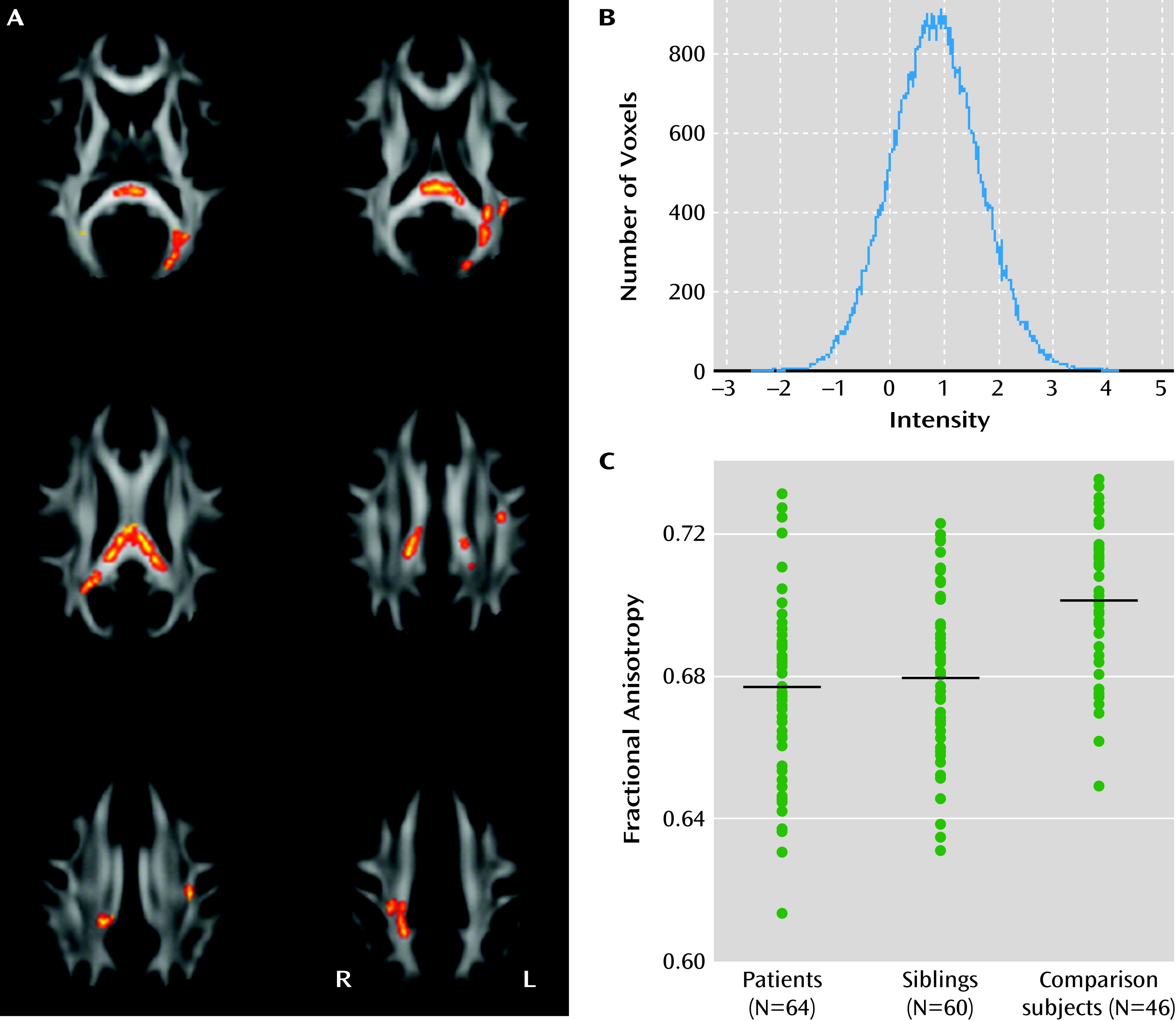
Comparing voxels within the significant case-comparison contrasts, we found reduced fractional anisotropy in unaffected siblings (p<0.05, family-wise error corrected; df=101) in the splenium and body of the corpus callosum, the posterior thalamic radiations, the posterior corona radiata, and the left superior longitudinal fasciculus (
Figure 2A and see Table S2 in the online
data supplement). We found no voxels in which anisotropy was significantly increased in siblings relative to comparison subjects. The histogram of the raw t statistic again showed a symmetric distribution, with the peak at t=0.86 (
Figure 2B).
Similar results were obtained when only right-handed individuals were included, when siblings with a history of depression (N=2) were excluded, when the two siblings with lowest fractional anisotropy were excluded, and when removing age, age squared, and sex covariates from the model.
In the siblings, region-of-interest analysis revealed a trend for reduced anisotropy in the forceps and the posterior thalamic radiations, which were not significant at 5% false discovery rate (
Table 2). No significant hemisphere-by-group interactions were found.
Average fractional anisotropy within the case-comparison cluster (
Figure 1A) for unaffected siblings was intermediate between the comparison group (t=3.32, p=0.001, df=105) and their affected siblings (t=3.21, p=0.0085, df=109;
Figure 1B).
Fractional anisotropy correlated highly between siblings with significant intraclass correlations (p<0.05, false discovery rate corrected) for most regions except the right retrolenticular internal capsule, the right cingulum, and the callosal tracts, which correlated at a trend level (
Table 3).
Associations With Clinical Measures
Duration of illness was negatively correlated with mean fractional anisotropy across the entire skeleton (rs=−0.41, p=0.0008) and with mean fractional anisotropy in the case-comparison cluster (rs=−0.54, p<10−5) and in the sibling-comparison cluster (rs=−0.41, p=0.0008). Age was negatively correlated with extracted fractional anisotropy measures (−0.16>rs>−0.33) and with duration of illness (rs=0.86). In a general linear model including age, age squared, and sex, the effect of duration of illness remained highly significant on mean fractional anisotropy across the skeleton (t=−2.93, df=58, p=0.0049), in the case-comparison cluster (t=−3.58, df=58, p=0.0007), and in the sibling-comparison cluster (t=−2.15, df=58, p=0.036), demonstrating that this relationship is largely independent of age. Adding a quadratic effect of duration of illness (t=1.70, p=0.09) did not change the above linear association between duration of illness and fractional anisotropy.
We found a significant effect of Lifetime Dimensions of Psychotic Symptoms scores on mean fractional anisotropy in the entire skeleton (t=−2.17, df=58, p=0.034), in the case-comparison cluster (t=−2.13, df=58, p=0.037), and in the sibling-comparison cluster (t=−2.66, df=58, p=0.010). This effect was independent of duration of illness, which did not correlate significantly with lifetime severity of psychotic symptoms.
Effects of Medication and Comorbid Disorders
We found no significant effects of antipsychotic, antidepressant, lithium, or mood stabilizer use on mean fractional anisotropy in the entire skeleton or within either of the clusters (in all cases, t<1.82, df=58, p>0.08). Similarly, diagnoses of past alcohol or substance abuse or dependence did not significantly affect fractional anisotropy (in all cases, t<0.91, df=167, p>0.36). Lifetime history of anxiety disorders was associated with reduced fractional anisotropy in the case-control cluster (t=2.15, df=167, p=0.03), but when covarying for anxiety disorders, the differences between patients and comparison subjects (t=4.77, df=104, p<10−5) and between siblings and comparison subjects (t=3.34, df=100, p=0.001) remained significant. Finally, current smokers had significantly lower mean fractional anisotropy within the significant case-control cluster (t=2.3, df=167, p=0.02). However, when adjusted for smoking status, fractional anisotropy within the case-control cluster remained significantly reduced in patients (F=5.36, df=104, p<10−6) and siblings (t=3.35, df=100, p=0.001). Together, these analyses demonstrate that medication use, substance abuse or dependence, and comorbid anxiety disorders alone cannot explain the observed group differences in fractional anisotropy.
Discussion
In the largest to date diffusion tensor imaging study of bipolar disorder to our knowledge, we found widespread white matter integrity reductions in patients that correlate with illness duration and severity of lifetime history of psychotic symptoms. These results are consistent with previous histological and imaging studies in bipolar disorder (
7,
14). We found similar, but more subtle, fractional anisotropy reductions in unaffected siblings. Fractional anisotropy values were also highly correlated between siblings, consistent with reports of high heritability for these measures (
9,
10). Together, these results suggest an important role for white matter integrity in the pathology of bipolar disorder and support the validity of white matter integrity as an endophenotype for the illness.
White matter structure is an attractive intermediate phenotype for bipolar disorders, and potentially the wider psychosis spectrum, for several reasons: white matter integrity is reliably measured using diffusion tensor imaging (
30); is directly related to relevant disconnectivity theories (
31,
32); continues to mature throughout adolescence and early twenties (
33), corresponding to the typical age of symptom onset; and is suggestive of molecular pathways putatively implicated in the disorder (
4,
34). Given the high heritability of fractional anisotropy (
9,
10) and our observations of reductions in unaffected relatives and high intraclass correlations between siblings, we postulate that a shared set of genes are responsible for white matter abnormalities and risk for bipolar disorder. This notion is supported by a twin study showing genetic correlations between bipolar disorder and total white matter volume (
35) and by decreased expression of white matter-related genes in patients with bipolar disorder (
3). More speculatively, the present findings are consistent with the notion that white matter integrity is a mediator in the effects of risk genes on the clinical phenotype. If so, investigating white matter integrity in molecular and cellular studies may provide new insights into downstream biological pathways and mechanisms of risk genes and may ultimately result in the identification of new medication targets. Alternatively, an increased frequency of risk alleles for other heritable conditions (e.g., hypertension) in bipolar patients may indirectly affect white matter integrity. Hence, additional study of gene identification and genetic effects is warranted to better understand the potential pleiotropy between bipolar disorder and white matter.
Although more than 25 studies have reported abnormal fractional anisotropy in bipolar patients, the locations of the reported effects are heterogeneous. Consistent with cognitive neuroscience theories of emotion and mood regulation (
32,
36), the most thoroughly described findings are within the frontal lobe and its connections with the medial temporal structures and the thalamus (
4,
7). Yet, there are as many reported white matter deficits outside these regions (
7,
14). In unaffected relatives, three studies showed localized effects (
11,
13,
14) and one indicated a global effect (
15). Our present voxel-wise and region-of-interest results also suggest widespread white matter perturbations in patients, while effects in the siblings were primarily restricted to the splenium of the corpus callosum, the posterior thalamic radiations, and the left superior longitudinal fasciculus. These results could reflect a true localization of familial influence in these regions, implying that fractional anisotropy reductions in other areas are determined by illness-specific factors. Alternatively, this apparent localization could reflect a statistical sensitivity bias favoring large, well-defined fiber bundles that enable the detection of subtler effects in unaffected siblings. Several observations are in favor of the latter interpretation, including those from a similarly large sample (
15). First, average fractional anisotropy extracted from the entire case-control cluster, which spanned more than half of the tract-based skeleton, was significantly reduced in siblings (
Figure 2). Second, the symmetrically rightward shift of the histogram of t statistics indicates a spatially consistent effect. Third, region-of-interest analysis revealed at least marginally reduced fractional anisotropy in all regions of interest except the anterior limbs of the internal capsule in unaffected relatives. Finally, histological data indicate that oligodendrocyte-related gene expression is highly homogeneous across the brain (
37), suggesting that a common set of genes influence white matter integrity throughout the brain. Thus, while our results suggest that the familial aspect of fractional anisotropy reductions is restricted to a set of specific tracts, we cannot rule out a more global effect masked by differential sensitivity across the white matter skeleton.
Of note, none of the studies reporting localized effects in unaffected relatives employed threshold-free cluster enhancement as was done here and in the 2011 study by Sprooten et al. (
15), despite it being more sensitive than other methods of voxel-wise statistics, particularly in the case of a subtle but spatially extended effect (
27). A lack of sensitivity to this type of signal could be an explanation for the spatial inconsistencies of previous studies, in addition to small sample size and heterogeneity.
We found highly significant associations between fractional anisotropy and duration of illness, implying a disease-specific component. These results could represent a differential aging or neurodegenerative aspect to the illness that would require longitudinal studies to further investigate (
38). Additionally, we show a negative association between severity of psychotic symptoms and fractional anisotropy, which is consistent with previous findings that history of psychosis in bipolar disorder is associated with abnormalities in prefrontal and fronto-limbic functional connectivity (
18). These clinical associations imply that more severe deficits in structural and functional connectivity may be responsible for the psychotic subtype of bipolar disorder.
A few methodological limitations should be mentioned. First, the unaffected siblings were mostly past the typical age of bipolar disorder onset. Therefore, apart from familial effects, the presence of protective factors in the unaffected siblings cannot strictly be ruled out. However, the unaffected siblings had fractional anisotropy values intermediate between the comparison subjects and patients, which seems incompatible with pronounced effects of resilience. Second, a sibling design cannot technically separate genetic effects from common environmental effects. Nevertheless, previous twin (
10) and pedigree (
9) studies have established that the common environmental component of fractional anisotropy is small to negligible. Third, regarding diffusion imaging, several experiments have shown that fractional anisotropy is sensitive to various types of pathology in white matter, including myelin damage, axonal disorganization, fiber incoherence, and packing density (
5,
6). Thus, while the interpretation of fractional anisotropy as a general index of white matter integrity has been validated, it cannot distinguish between specific microstructural detriments. Finally, tract-based spatial statistics optimize registration to white matter anatomy, allowing unbiased voxel-wise comparisons without a priori region selection, but they remain sensitive to registration errors. To localize results to specific tracts, complimentary methods such as tractography are desirable.
In summary, the present data confirm significant global fractional anisotropy reductions in affected individuals and more subtle reductions in unaffected relatives. Fractional anisotropy within tracts of interest and within significant clusters correlated highly between siblings, supporting a significant underlying familial component. Overall, these results confirm that white matter plays an important role in bipolar disorder and encourage its use as an endophenotype for genetic studies of the disorder.