Suicide, with a worldwide annual mortality approaching 1 million, is anteceded by suicidal ideation—thoughts and wishes to kill oneself (
1,
2). Although suicidal ideation leads to completed suicide only rarely (
2,
3), it cannot be ignored clinically (
1,
2), and the management of severely suicidal outpatients remains an enormous clinical challenge (
2,
4). Standard antidepressants relieve suicidal ideation, but this may take several weeks, and not all patients respond adequately (
1,
4–
7). Atypical antipsychotics (
8,
9) and lithium (
10) have been found to decrease suicidal ideation and/or suicidal behavior in specific patient populations. Ketamine is effective as a quick-acting treatment for suicidal ideation and depression, but necessitates repeated administration under medical supervision (
11,
12). Thus, no short-term pharmacological treatments that are suitable for independent outpatient use are currently available for suicidal ideation.
Depression, which is often accompanied by suicidal ideation, shares neurobiological and psychological characteristics with separation distress—the innate, painfully anxious, dysphoric response of young animals and humans to separation from their attachment figures (
13–
17). Remarkably low, nonsedating doses of opioid analgesics have been found to inhibit separation distress in every animal species tested (
15–
17). The neuroanatomy of the separation distress system overlaps with the brain’s “pain matrix” and shares some of its neurotransmitter substrates (
16,
17). It has been suggested that abrupt cessation of opioid release upon separation from attachment figures contributes to painful separation feelings in animals and humans (
16,
17).
Patients with borderline personality disorder, who are exquisitely sensitive to separations, often become suicidal after interpersonal rejections (
4,
18), and this patient population has been found to have abnormalities in their endogenous opioid systems (
19). Among patients in psychotherapy, most suicidal acts occur after interpersonal losses or rejections (
20), and analgesic treatment has been shown to decrease social pain in rejected lovers (
21). Social rejection activates the endogenous opioid system in healthy volunteers (
22,
23), and this activation is altered in depressed patients (
24).
Opioids were widely used to treat depression from about 1850 to 1956 (
25). Because of their addictive potential and lethality in overdose, opioids were replaced by standard antidepressants once these became available. However, several studies since then have found them to be effective for treating depression (
25–
27).
In addition to their association with depression and separation distress, physical and mental pain are associated with suicidality (
28–
31). Some studies have found mental pain to be the psychological variable most strongly associated with current suicidality, more so than depression and hopelessness (
28–
30).
In sum, converging lines of evidence point to a connection between separation distress, mental pain, depression, suicidal ideation, and endogenous opioids (
14–
31). It was therefore hypothesized that opioids in very low dosages might help alleviate suicidal ideation.
Buprenorphine, which is utilized in high dosages for the treatment of opioid use disorder, has a complex pharmacological profile. It is a partial mu agonist and a potent kappa antagonist, and it has several active metabolites (
32). Its kappa antagonism may enhance its antidepressant action (
16,
27). It causes much less respiratory depression than other opioids and is therefore safer in overdose (
32,
33). We aimed to assess the efficacy and safety of ultra-low-dose buprenorphine as a time-limited adjunctive treatment for suicidal ideation.
In animal studies, morphine at 0.125 mg/kg, less than half the dose used for analgesia (
34), has been shown to cause potent inhibition of separation distress vocalizations (
17). To our knowledge, this is the lowest dose found to produce a robust behavioral response in animals. Given the high sensitivity of the mammalian separation distress system to opioids, we used minimal, subanalgesic dosages of buprenorphine (e.g., 0.1 mg once or twice daily), which were gradually increased. This approach utilized the lowest possible dosages of a potentially addictive drug and minimized side effects that might reduce adherence, increase dropout, and unblind study drug assignment.
Opioids are involved in more deaths than any other drug class in fatal pharmaceutical overdoses in the United States (
35). Thus, the lower lethality of buprenorphine and the very low dosages employed in this study were crucial for enabling its independent, home-based use. However, buprenorphine is potentially addictive and possibly lethal (
32). We therefore designed this study as a time-limited trial for severely suicidal patients without substance abuse. A treatment duration of 4 weeks was chosen because it roughly corresponds to the interval between the start of antidepressant treatment and the onset of clinical improvement (
5–
7), and in order to minimize the risk of developing dependence.
Method
Study Design and Participants
We enrolled patients from four medical and psychiatric centers in Israel: Abarbanel Mental Health Center, Bat Yam; Hadassah Medical Center, Jerusalem; Jerusalem Mental Health Center, Jerusalem; and Wolfson Medical Center, Holon. Recruitment was conducted from January 2010 to July 2013. Patients were eligible to participate if they were between 18 and 65 years of age and suffered from clinically significant suicidal ideation, as indicated by a score ≥11 on the self-report version of the Beck Scale for Suicide Ideation (
36,
37) for at least 1 week. Exclusion criteria were a lifetime history of opioid abuse, a lifetime diagnosis of schizophrenia, current psychosis, ECT within the past month, substance or alcohol abuse within the past 2 years, and benzodiazepine dependence within the past 2 years. Pregnant or lactating women and patients who suffered from any severe medical condition were also excluded. Psychiatric diagnoses were made according to DSM-IV-TR criteria. The study protocol was approved and monitored by the institutional review boards of all participating centers. All participants provided written informed consent before entering the study.
Randomization
Within each center, randomization was done in a double-blind manner with a web-based schedule (
www.randomizer.org), with buprenorphine and placebo assigned in a 2:1 ratio. Sublingual gelatin-based lozenges of 0.1 mg of buprenorphine and identical placebo lozenges were dispensed by pharmacies at each participating center. All study personnel and participants were unaware of participants’ treatment assignment for the duration of the study.
Procedures
Questionnaires were administered once a week. Study psychiatrists met with all participants at baseline and once a week during the trial. At each visit they assessed the severity of participants’ suicidality, screened them for adverse events, and adjusted their medication dosages. An additional appointment with the study psychiatrists was made 1 week after discontinuation of the study medication to screen for possible withdrawal symptoms and verify transition to ongoing care.
Sublingual buprenorphine lozenges were administered on a flexible schedule, beginning with 0.1 or 0.2 mg/day. Once a week, at the decision of the study psychiatrists, the daily dose could be raised in 0.1–0.2 mg increments, to a maximal daily dose of 0.8 mg. The dose was not raised if participants were found to have reached full remission (i.e., had a score of zero on items 4 and 5 of the Beck Scale for Suicide Ideation) or if they experienced significant adverse events. A week’s supply of medication (≤5.6 mg, usually ≤2.8 mg) was not considered to present a high risk for suicide by overdose. Outpatients received the study medication for the following week during their weekly visits, and took it independently at home. Average adherence, measured by pill counts, was 92%. Inpatients received the study medication daily from their hospital staff. Average medication dosages during the trial are summarized in Figure S1 in the data supplement that accompanies the online edition of this article.
Outcome Measures
The primary outcome measure was change from baseline in score on the Beck Scale for Suicide Ideation (
36) at the end of the study. Secondary outcome measures were change from baseline in scores on the Beck Depression Inventory II (
38) and the Suicide Probability Scale (
39) and its hopelessness and worthlessness factors. Mental pain does not have a universally accepted definition (
29). Changes in its level were assessed by item 25 of the Suicide Probability Scale (“I feel it would be less painful to die than to keep living the way things are”).
Safety
We consulted and received permission from the Israel Ministry of Health, Division of Clinical Studies, to carry out this trial. Participants’ recruitment was contingent on their being in treatment with a mental health professional, clinic, or hospital that was not part of the study team. In addition to the written informed consent we obtained from all participants, we obtained the approval and collaboration of their treating clinicians. Given the ethical considerations, the study was designed as an adjunctive trial; more than 70% of the participants were on antidepressants, and almost all took some psychotropic medication other than the study drug. With the exception of antidepressants, the treating clinicians could modify the pharmacotherapy their patients were receiving, and could see them as frequently as they deemed necessary, in addition to their weekly appointments with the study psychiatrists. Participants on antidepressants had to have been taking them for at least 28 days, and no changes were allowed during the study period. Table S1 in the online data supplement lists all psychotropic medications taken by study participants during the study. There were no completed suicides during the study period.
Statistical Analysis
To the best of our knowledge, no controlled trial has tested the effects of buprenorphine on suicidal ideation. Based on results from a study on the antisuicidal effects of risperidone (
9), we calculated that a sample of 75 participants with a 2:1 randomization ratio would provide 80% power at a 5% significance level to detect a 6.3-point difference between groups in scores on the Beck Scale for Suicide Ideation, assuming a standard deviation of 9.0.
We assessed treatment efficacy with modified intent-to-treat analyses, which included data from all participants who received at least one dose of study drug and had at least one suicidal ideation measurement after baseline. Sensitivity analyses evaluated the robustness of conclusions in relation to missing data. Mixed models for repeated-measures analyses were used to determine the effects of treatment on change from baseline in score on the Beck Scale for Suicide Ideation. The model included treatment group, medication dosage, scheduled visit, treatment-by-visit interaction, baseline score on the Beck Scale for Suicide Ideation, current major depressive episode, and current major depressive episode-by-visit interaction. Missing values were not imputed. An unstructured covariance structure was used.
Secondary outcome analyses, sensitivity analyses, and post hoc baseline-to-endpoint primary outcome analyses were done with and without last observation carried forward. Endpoints were changes from baseline to the end of week 4. We used an analysis of covariance model that included baseline assessment score, treatment group, study medication dosage, and current major depressive episode.
Treatments were compared with two-sided t tests for continuous variables and with Pearson’s chi-square test or Fisher’s exact test, as appropriate, for categorical variables. Analyses were conducted with SPSS, version 20 (IBM, Inc., Armonk, N.Y.). Correlations were calculated using Pearson’s r. Fisher’s r-to-z transformation (
http://vassarstats.net/) was used for testing the difference between correlations. All comparisons were two-sided. A p value <0.05 was regarded as significant for all analyses. Safety analyses were done on the safety population, defined as all randomly assigned participants who took at least one dose of study drug, and are presented as descriptive statistics.
Results
Participants
Of 265 patients screened, 88 were randomly allocated to treatment groups—57 to the buprenorphine group and 31 to the placebo group (for a CONSORT diagram, see Figure S2 in the online
data supplement). Of these, 62 (70.5%) received at least one dose of study drug and had a baseline score and at least one postbaseline score on the Beck Scale for Suicide Ideation.
Table 1 summarizes the baseline demographic and clinical characteristics of the two treatment groups; there were no significant differences between groups.
Participants in this study were severely suicidal (average baseline score on the Beck Scale for Suicide Ideation, 19.7), and 64.5% of them had made at least one suicide attempt. More than half (56.8%) met criteria for borderline personality disorder. Almost all were clinically unstable, and their ability to cooperate with the study team was compromised, as reflected in a high dropout rate (29.5%) during the first week of treatment.
Primary Outcome Measure
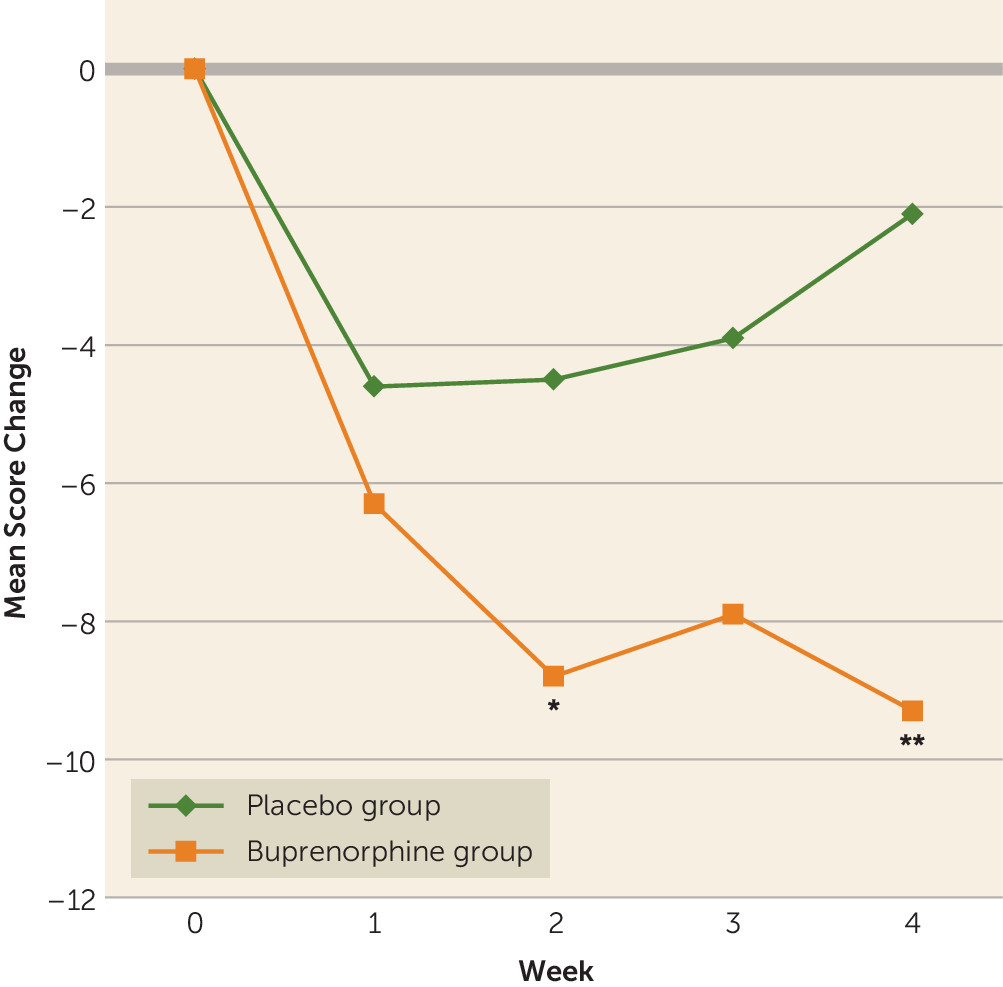
Patients in the buprenorphine group had a greater reduction in Beck Suicide Ideation Scale score than patients in the placebo group, both at the end of week 2 (mean difference=−4.3, 95% CI=−8.5, −0.2; p=0.04) and at the end of week 4 (mean difference=−7.1, 95% CI=−12.0, −2.3; p=0.004) (
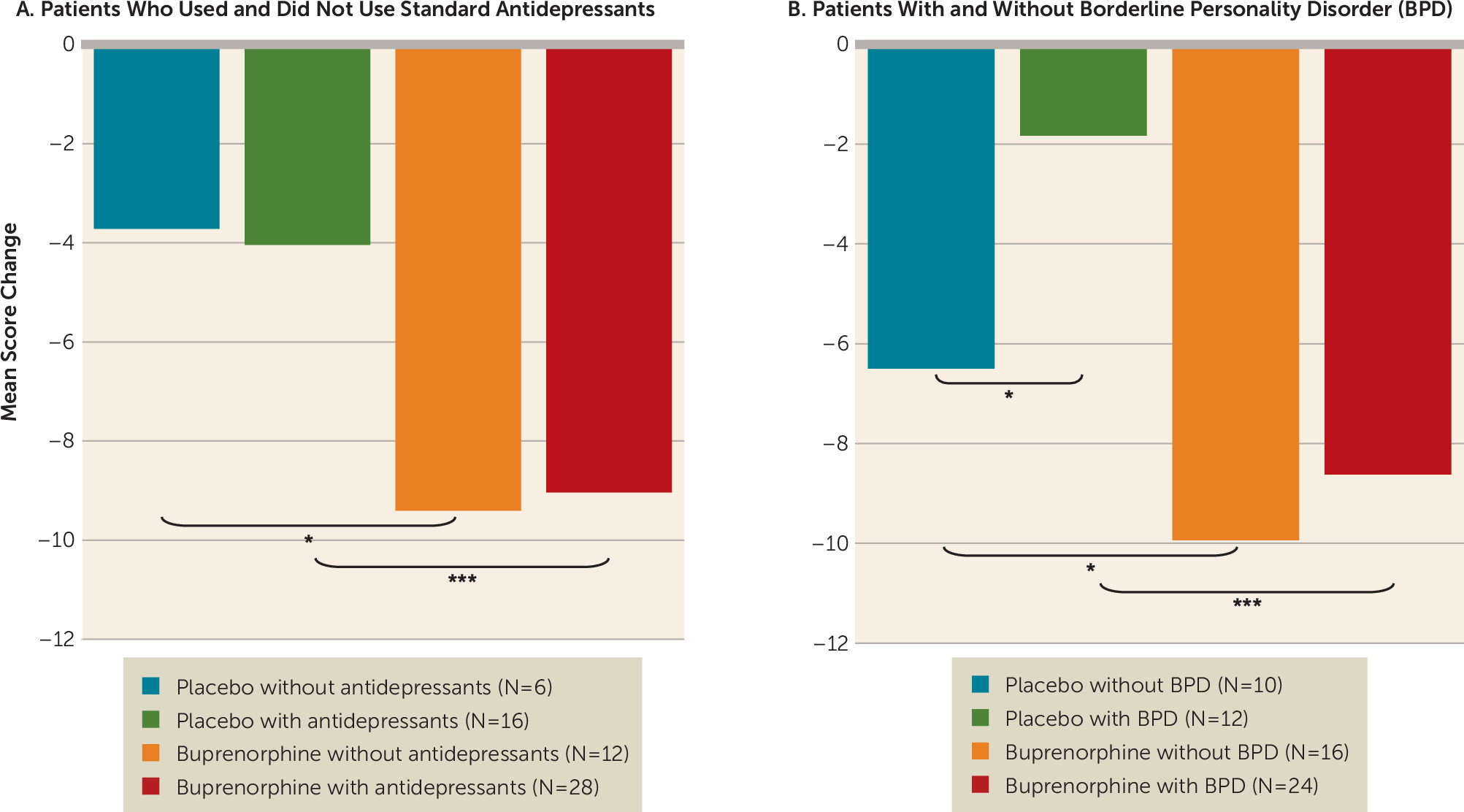
Figure 1). Sensitivity analysis revealed similar results (mean difference=−5.7, 95% CI=−10.1, −1.4; p=0.01). Concurrent treatment with antidepressants did not affect this response pattern: post hoc comparisons by a series of independent t tests on endpoint results found a greater reduction in Beck Suicide Ideation Scale score in participants who received buprenorphine compared with those who received placebo, whether or not they were also treated with standard antidepressants (
Figure 2A). While a diagnosis of borderline personality disorder was associated with a lower response to placebo, it did not attenuate the response to buprenorphine: post hoc comparisons found a greater reduction in Beck Suicide Ideation Scale score in participants who received buprenorphine compared with those who received placebo, whether or not they also had a diagnosis of borderline personality disorder (
Figure 2B).
Secondary Outcome Measures
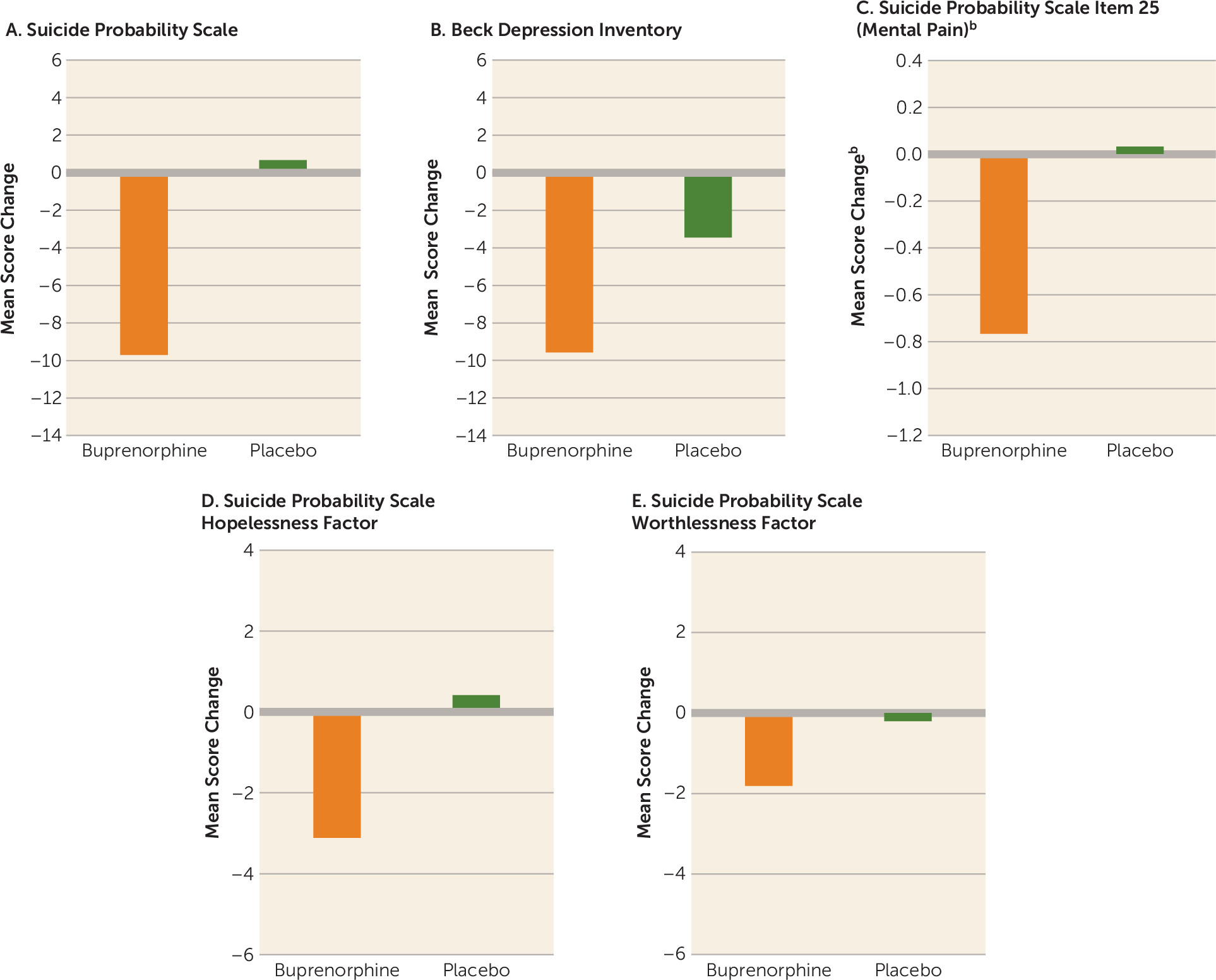
Patients in the buprenorphine group had a greater reduction in score on the Suicide Probability Scale, an independent measure of suicidal ideation, compared with those who received placebo (mean difference=−10.4, 95% CI=−19.7, −1.0; p=0.03) (
Figure 3A). When the analysis was restricted to patients who completed the study (i.e., without last observation carried forward), the scores of the buprenorphine group were lower still, although the difference now fell short of significance (mean difference=−12.0, 95% CI=−24.3, 0.3; p=0.055).
Patients in the buprenorphine group had lower Beck Depression Inventory scores than those in the placebo group, although the difference did not reach significance (mean difference=−6.1, 95% CI=−13.2, 1.0; p=0.09) (
Figure 3B). Restricting the analysis to patients who completed the study yielded similar results (mean difference=−8.3, 95% CI=−16.7, 0.1; p=0.052). Overall, the magnitude of reduction in depressive symptoms was smaller than the reduction in suicidal ideation (see Figure S3 in the online
data supplement). In post hoc analyses, patients who met criteria for major depressive disorder had a lower rate of placebo response and smaller reductions in depressive symptoms compared with nondepressed patients, but these differences were not significant (see Figure S4 in the
data supplement).
Patients in the buprenorphine group had a reduction in level of mental pain, as assessed by item 25 of the Suicide Probability Scale (mean difference=−0.8, 95% CI=−1.5, −0.7; p=0.03) (
Figure 3C). Restricting the analysis to patients who completed the study yielded a similar effect (mean difference=−0.9, 95% CI=−1.8, −0.02; p=0.045). Patients in the buprenorphine group had lower scores than those in the placebo group on the hopelessness factor of the Suicide Probability Scale, although the difference fell short of significance (mean difference=−3.5, 95% CI=−7.3, 0.3; p=0.07) (
Figure 3D). Differences in changes in the worthlessness factor of the Suicide Probability Scale were not significant (
Figure 3E).
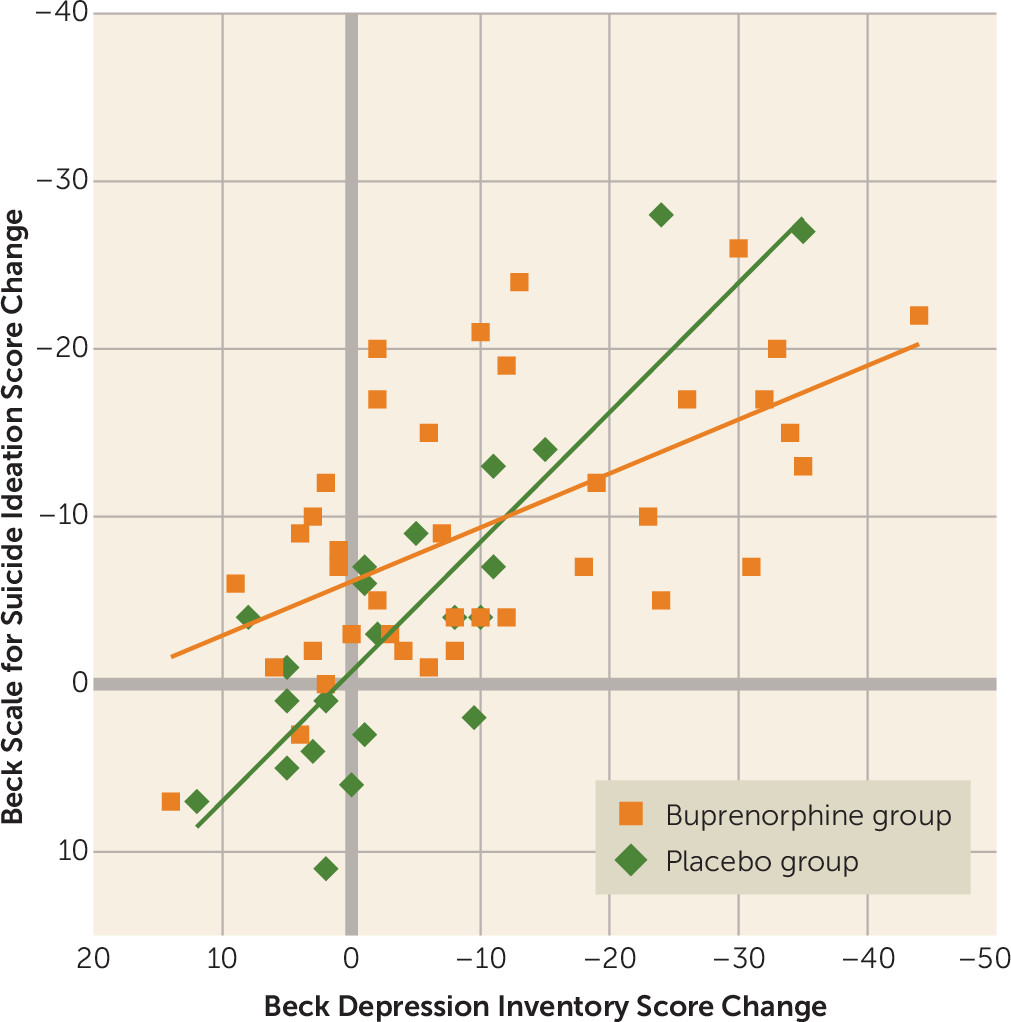
The correlation between individual Beck Suicide Ideation Scale and Beck Depression Inventory score changes from baseline to week 4 was strong in both groups, but stronger in the placebo group (r=0.85, 95% CI=0.7, 0.9; p<0.001) compared with the buprenorphine group (r=0.58, 95% CI=0.3, 0.8; p<0.001) (for the difference between correlations, p=0.04) (
Figure 4). This difference raises the possibility that treatment with ultra-low-dose buprenorphine may have decreased the tight coupling between changes in depressive and suicidal symptoms. Restricting the analysis to patients who completed the study yielded similar correlations (placebo: r=0.85, 95% CI=0.6, 0.9; p<0.001; buprenorphine: r=0.59, 95% CI=0.3, 0.8; p<0.001); the difference between correlations fell short of significance (p=0.08).
Adverse Events
One or more adverse events were reported in 77.2% of participants in the buprenorphine group and 54.8% of those in the placebo group (p=0.03). Among participants in the buprenorphine group, there were more reports of fatigue (49.1% compared with 22.6% in the placebo group), nausea (36.8% compared with 12.9%), dry mouth (29.8% compared with 9.7%), and constipation (26.3% compared with 9.7%). (For frequencies of all adverse events in each group, see Table S2 in the online data supplement.) Despite the high incidence of adverse events, and considering the overall compromised adherence of the study population, the treatment was well tolerated: discontinuation due to adverse events occurred in 22.8% of those who received buprenorphine and in 16.1% of those who received placebo (n.s.). The most common adverse events leading to study discontinuation were nausea, headache, and dizziness.
One or more serious adverse events occurred in 5.3% of the participants who received buprenorphine and in 6.5% of those who received placebo (n.s.). Two participants (one each in the buprenorphine and placebo groups) made a suicide attempt during the study period. One of them was hospitalized for psychiatric observation.
At the end of week 4, the study medication was discontinued without a taper. All participants denied withdrawal symptoms during their follow-up appointment 1 week later. It is possible that in this opioid-naive population, the short duration and low dosages protected against dependence. Alternatively, symptoms may have emerged during the first few days after discontinuation and abated by the time of the follow-up appointment. Among study completers, no exacerbation in suicidality was reported 1 week after medication discontinuation, but clearly a longer follow-up period is required to assess this.
Discussion
In this double-blind, placebo-controlled trial, very low dosages of buprenorphine were associated with decreased suicidal ideation in a group of severely suicidal patients. The choice of ultra-low-dose buprenorphine as a treatment for suicidal ideation was theory driven, based on human and animal studies that have linked suicidality with mental pain and endorphinergic control of the separation distress system (
14–
31).
With a few notable exceptions (
4,
8,
11), pharmacological approaches to the treatment of suicidality have viewed it as an aspect of treating depression (
1,
5). The relationship between suicidality and depression is complex (
1,
2,
5,
6), and our findings in this study appear to support the view that suicidal ideation and depression may be distinct, although related, phenomena, at least with respect to their response to pharmacotherapy. While higher dosages of buprenorphine than those we used have been found to exert a strong effect on depressive symptoms (
26,
27), our findings are in agreement with those of Ballard et al. (
11), who found that improvements in suicidal ideation after ketamine infusion were related to, but not completely driven by, improvements in depression and anxiety. Indeed, it was recently suggested that some of ketamine’s psychotropic effects may be mediated by its mu agonism (
40).
Two somewhat unexpected findings of this trial require corroboration and further study. First, the effects of ultra-low-dose buprenorphine on suicidal ideation did not differ between patients who were concomitantly treated with antidepressants and those who were not. Second, unlike most antidepressant medication trials, in which a diagnosis of borderline personality disorder has been associated with a poorer clinical outcome (
4), patients with and without borderline personality disorder in this trial had similar responses to buprenorphine. This finding, if replicated, raises the possibility that ultra-low-dose buprenorphine treatment addresses a subset of affective symptoms—those associated with painful feelings of rejection and abandonment, and that in this dosage range, it is less active against neurovegetative and other symptoms that are more related to reduced hedonic tone (i.e., in Panksepp’s terminology, ultra-low-dose buprenorphine treatment may attenuate the hyperactivation of the endorphinergic
panic/grief system, without reversing the partial shutdown of the aminergic
seeking system) (
15,
16). As a speculative corollary, in this dosage range, buprenorphine might be more effective against “atypical/borderline” suicidality than against “melancholic” suicidality.
This study had several important limitations. First, its outcome measures were based on self-scored questionnaires. While suicidal ideation is inherently a personal, subjective experience, cross-validation by clinician-scored scales of suicidality, depression, and overall functioning would be beneficial and should be part of any future trial. Second, study participants came from an unstable, severely suicidal population, and a majority of them met criteria for borderline personality disorder. Their ability to comply with the protocol was compromised, as evidenced by relatively high dropout rates in both the placebo and buprenorphine groups. It remains to be seen whether the findings of this study are applicable to more stable, less severely suicidal patients. Third, the dosing regimen of the study medication was flexible and gradual, limiting the inferences that may be drawn about the optimal dosage of buprenorphine to treat suicidal ideation. Fourth, the diagnostic, pharmacological, and therapeutic heterogeneity of the study population and its relatively modest size limited our ability to stratify results by dosage, site, gender, diagnosis, and other potentially important factors. Fifth, this study did not assess nonsuicidal self-injury, which is associated with borderline personality disorder, mental pain, and abnormalities in the endogenous opioid system (
23). Sixth, the trial did not include an extended follow-up period, which would have allowed assessment of possible long-term effects, both positive and negative, of this short-term treatment, including the possibility of developing drug craving or rebound suicidality. More research is needed to address all these deficiencies.
Finally, despite its favorable safety profile, buprenorphine is potentially addictive and possibly lethal (
32). In our opinion, even in the very low dosages employed in this trial, it should be tested only in individuals who have been screened for the possibility of substance abuse. This is a single, time-limited trial of an experimental treatment for suicidal ideation rather than suicidal behavior, and its results do not support the widespread, long-term, or nonexperimental use of buprenorphine for suicidality.
Conclusions
Human and animal studies have found many links between suicidal ideation, mental pain, depression, and opioid regulation of neurotransmission in the separation distress system (
14–
31). In this study, the time-limited use of very low dosages of buprenorphine was associated with a decrease in severe suicidal ideation. Further research with longer follow-up is required to establish the safety, dosing, and appropriate patient populations for this experimental short-term treatment.
Acknowledgments
The authors thank their study participants; their colleagues Dr. O. Boneh, Dr. E. Danilovich, Dr. D. Gofrit, Dr. M. Gonzalves, Dr. R. Idelman, Dr. M. Lipot, Dr. H. Oren, Dr. T. Pomerantz, Dr. M. Swartz, Dr. O. Yatziv, and Dr. Y. Zimmerman, who assisted them in this study; L. Kogan, R. Sadan, K. Samet, O. Schnabel, T. Shahar, and L. Yossefi for their help with data collection; and E. Zur, of Super-Pharm Professional Laboratories, for his help in preparing the study medications.