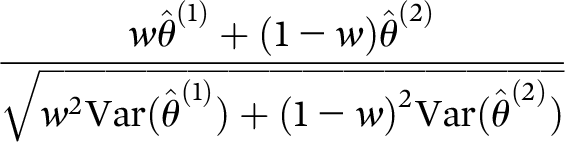An estimated 60%–70% of patients with major depressive disorder have an inadequate response to initial treatment (
1–
3), resulting in substantial persistent impairment and high utilization of health care resources (
4). While therapeutic strategies such as switching antidepressants and using adjuvant drug treatments can improve response, almost 40% of patients remain symptomatic and fail to achieve full remission (
5,
6). There is an urgent public health need for alternative antidepressant treatments.
Opioids have been used for centuries to treat what are now recognized as mood disorders and were recommended for melancholia in the 19th and early 20th centuries until they were displaced in the 1950s by monoamine-based antidepressants (
7,
8). Since that time, the monoamine hypotheses for the pathophysiology of depression notwithstanding, a growing body of preclinical and clinical evidence supporting the hypothesis that mood disorders involve dysregulation of the endogenous μ- and κ-opioid system has emerged (
7,
9,
10). Imaging studies have revealed alterations in opioid transmission associated with induced sadness, social rejection, and depression in key brain emotional regulatory centers, including the nucleus accumbens, ventral pallidum, amygdala, anterior cingulate cortex, and posterior thalamus (
11–
13). Furthermore, there is evidence of endorphin deficiency in severely depressed and suicidal patients (
12,
14,
15). Additionally, uncontrolled clinical studies have described the efficacy of low-dosage treatment with opioids, including buprenorphine, for treatment-refractory depression (
8,
16), whereas treatment with a μ-opioid antagonist had no effect (
17). Interestingly, there is epidemiologic evidence that chronic high-dosage opioid agonist therapy may increase the risk of depression (
18).
Despite substantial gains in our understanding of the role of endogenous opioid dysregulation in the context of depression and the need for antidepressants with alternative mechanisms of action, contemporary clinical use of opioid agonists as antidepressants in clinical practice remains highly limited because of unresolved issues of abuse and dependence. To overcome the limitations of opioid agonists, we developed ALKS 5461—a combination of buprenorphine, which is a partial μ-opioid receptor agonist (
19), and samidorphan (previously referred to as ALKS 33), which is a potent μ-opioid receptor antagonist (
20), formulated in a single sublingual tablet. Because of high first-pass hepatic metabolism, buprenorphine is generally administered sublingually. Samidorphan was included in the combination to block the μ agonist effects of buprenorphine associated with its abuse and addictive potential. Samidorphan has high sublingual bioavailability and was thus suitable for coformulation with buprenorphine in a single tablet.
In addition to its effects on μ-opioid receptors, buprenorphine has also been shown in vivo to block the action of κ-opioid agonists (
21) and has been characterized in vitro to be a partial κ agonist with low intrinsic activity (
19). The buprenorphine/samidorphan combination results in high-affinity binding with net low intrinsic activity at both μ- and κ-opioid receptors. It is hypothesized that the combination serves to decrease or dampen opioid tone in regions of excess endogenous μ- and κ-opioid ligand activity and restore opioid tone in regions where such activity is impaired.
Previous studies with the buprenorphine/samidorphan combination systematically evaluated relative ratios of its components to block the opioid abuse liability signal (
22). A 1:1 ratio of buprenorphine to samidorphan was found to maximally block subjective and objective measures of μ-opioid activity, including pupillary effects, subjective drug liking, and euphoria. A subsequent small 1-week pilot study characterized the efficacy signal of an 8 mg/8 mg dosage of buprenorphine/samidorphan after 1 week of adjunctive treatment in patients with major depression who had an inadequate response to standard antidepressants (
22). The present study was undertaken to evaluate the efficacy and tolerability and to preliminarily characterize an active dosage range of a 1:1 ratio of buprenorphine to samidorphan over 4 weeks as adjunctive therapy in patients with major depression who had an inadequate response to standard antidepressants. The 8 mg/8 mg dosage was selected to expand the findings from the pilot study (
22), and the 2 mg/2 mg dosage was selected to determine whether a lower dosage would also yield antidepressant activity.
Clinical trials in depression have been afflicted by increasing rates of placebo response (
23). High placebo response interferes with assessment of efficacy and masks treatment effects. In order to address excessive placebo response, this trial employed a sequential parallel comparison design. This design was first proposed for use in depression clinical studies (
24) to enhance signal detection by reducing placebo response while increasing study efficiency (
25,
26). The sequential parallel comparison design is a two-stage study design. Stage 1 is a double-blind placebo-controlled parallel comparison with a higher proportion of patients randomized to placebo than to active drug. Patients randomized to placebo who meet criteria for placebo nonresponse in stage 1 are rerandomized in a blinded fashion to active drug or placebo in stage 2. The prespecified primary efficacy evaluation of a sequential parallel comparison design is based on a weighted combination of test statistics from all patients in stage 1 and the rerandomized placebo nonresponders in stage 2.
Method
The study was conducted at 31 sites in the United States between December 2011 and March 2013 in accordance with the Declaration of Helsinki (1964) and Good Clinical Practice principles outlined in the International Conference on Harmonization (1997). The protocol, amendments, and informed consent were approved by an institutional review board for each site, and written informed consent was obtained from all participants.
Study Design
This randomized, double-blind, placebo-controlled study utilized a two-stage sequential parallel comparison design. Each 5-week stage consisted of a 4-week treatment period followed by a 1-week taper. In stage 1, patients were randomized in a 2:2:9 ratio to receive buprenorphine/samidorphan at 2 mg/2 mg (the 2/2 dosage group) or 8 mg/8 mg (the 8/8 dosage group) or placebo. This stage 1 randomization ratio was anticipated to result in an approximate placebo nonresponder yield of 65%, defined as a <50% reduction in score on the 17-item Hamilton Depression Rating Scale (HAM-D) (
27) in stage 1 and a baseline score >14. This resulted in a number of placebo nonresponders considered appropriate for 1:1:1 randomization to the 2/2 dosage, the 8/8 dosage, or placebo in stage 2. The stage 1 placebo responders continued on placebo in stage 2. All patients continued on their current antidepressant therapy and on the same dosage throughout the course of the study (see Table S1 in the
data supplement that accompanies the online edition of this article). The placebo or buprenorphine/samidorphan study drug was administered daily as sublingual tablets matched in size and shape. Patients assigned to the 8/8 dosage group were initiated with the 2/2 dose on day 1 and their dosage was increased to the 8/8 dosage over 3 days.
Trained and certified site-based raters administered both the HAM-D and the Montgomery-Åsberg Depression Rating Scale (MADRS) at each study visit. To manage the potential for site-based rater bias and to verify rating reliability, site-based HAM-D and MADRS interviews were recorded and submitted for site-independent central scoring. Site-independent and site-based ratings were compared for quality control.
Patient Selection
Men and women 18–65 years of age with a BMI ≤40 were eligible if they met Structured Clinical Interview for DSM-IV-TR Axis I Disorders criteria for a diagnosis of major depressive disorder, which was validated using a SAFER interview administered by remote, independent raters (
28). Inclusion criteria were a current episode of major depression for ≤24 months; a HAM-D score ≥16 at screening; treatment with a selective serotonin reuptake inhibitor (SSRI) or a serotonin-norepinephrine reuptake inhibitor (SNRI) at an adequate dosage (as defined by the Massachusetts General Hospital Antidepressant Treatment Response Questionnaire) for at least 8 weeks; and an inadequate response to one or two courses of an antidepressant (defined as a <50% response on the Antidepressant Treatment Response Questionnaire [
29], which was independently confirmed by remote, independent raters during the SAFER interview). Participants had to be otherwise healthy based on physical examination, history, 12-lead ECG, and laboratory tests.
Exclusion criteria were psychotic symptoms during the current episode; a decrease of >25% or ≥8 points in HAM-D score from screening to the baseline visit; initiation of psychotherapy within 6 weeks of screening; use of other adjunctive therapy during the current episode; use of opioid agonists or naltrexone within 2 months before screening; ECT during the current episode; suicide attempt within the past 2 years; a history of alcohol or substance dependence within the past 12 months; any lifetime history of opioid dependence; or a positive drug test. Women who were pregnant, planning to become pregnant, or breastfeeding were excluded.
Study Assessments
Efficacy.
The primary outcome measure was the change in HAM-D score (
30) from baseline to the end of the 4-week treatment period. Secondary outcome measures were the change from baseline to the end of the 4-week treatment period in MADRS score (
31) and Clinical Global Impressions severity scale (CGI-S) score (
32), rate of response (defined as a reduction ≥50% in HAM-D or MADRS score from baseline to week 4), and rate of remission (defined as a HAM-D score ≤7 or a MADRS score ≤10 at week 4). Exploratory outcome measures were scores on the Inventory of Depressive Symptomatology–Self-Report (
5), the Sheehan Disability Scale (
33), and the 12-item Short-Form Health Survey, Version 2 (SF-12) (
34). Efficacy was assessed weekly throughout the course of the study.
Safety and tolerability.
Treatment-emergent adverse events, ECG, vital signs, and laboratory evaluations were monitored throughout the study. Analysis of treatment-emergent adverse events was conducted across stage 1 and stage 2. Patients receiving different treatments in stage 1 and stage 2 were included in the analysis of both treatment groups. Attribution of a particular treatment-emergent adverse event was based on the treatment the patient was receiving at the time of the adverse event’s onset.
Visual analogue scales for drug liking and subjective effects were used to assess subjective effects; the Clinical Opiate Withdrawal Scale (
35) was used to assess opioid withdrawal symptoms; the Addiction Research Center Inventory–Morphine Benzedrine Group scale (
36) was used to assess drug-induced mood effects; and the Columbia–Suicide Severity Rating Scale (C-SSRS) (
37) was used to assess suicidal behavior and ideation.
Urine drug screens to detect illicit substances were conducted at screening, baseline, and end of study, testing for amphetamines, barbiturates, benzodiazepines, cocaine, tetrahydrocannabinol, methadone, opioids, phencyclidine, and, at screening only, buprenorphine.
Statistical Analysis
The efficacy populations in each stage included all randomized patients who received at least one dose of study drug and had at least one postbaseline efficacy assessment. The safety population included all patients who received at least one dose of study drug.
The primary efficacy endpoint, HAM-D score change from baseline to the end of the 4-week treatment period, was evaluated using the weighted combination of statistics from the stage-specific mixed models for repeated measures (MMRM). The models included variables for treatment group, visit, treatment-by-visit interaction term, and baseline HAM-D score and specified a first-order autoregressive covariance matrix. The Kenward-Roger approximation (
38) was used to adjust the denominator degrees of freedom. The treatment effect was assessed as the differences in least-squares mean change from baseline to week 4 for the buprenorphine/samidorphan and placebo groups, combined across stages 1 and 2 using prespecified 0.6/0.4 weighting for stage 1/stage 2. Sensitivity analysis was also conducted using 0.5/0.5 weights. Combined inference was conducted using the weighted linear combination of stage-wise test statistics:
where
w=0.6.
In simulation sequential parallel comparison design studies using MMRM, stage 1/stage 2 weights of 0.6/0.4 resulted in a robust test statistic that demonstrated type I error control, optimal power, and minimum bias under the assumption of missing at random (
39). Sensitivity analysis using weights of 0.5/0.5 resulted in a study treatment effect slightly greater than when using the prespecified weights of 0.6/0.4, resulting in unchanged interpretation and conclusions. The assumption of missing at random in clinical trials of major depression is supported by a meta-analysis of 25 New Drug Application data sets reported by the U.S. Food and Drug Administration. The same statistical methods were used to evaluate other continuous variables based on change from baseline.
Rates of treatment response and remission were calculated as the number of patients meeting the response and remission criteria, respectively, out of the total number of patients who had a HAM-D or MADRS assessment at week 4. Exact confidence intervals were calculated.
A total sample size of 130 patients was estimated to provide at least 85% power at a two-sided significance level of 0.05 assuming a mean change in HAM-D score of −7.0 (SD=7.0) and −10.5 (SD=7.5) in stage 1, and −3.0 (SD=5.3) and −7.0 (SD=6.5) in stage 2 for the placebo and buprenorphine/samidorphan groups, respectively.
Missing data were handled using MMRM’s maximum likelihood rather than an imputation method.
Results
The sample’s baseline demographic and clinical characteristics, summarized in
Table 1, did not differ significantly between groups for stage 1 or stage 2. At initial randomization, 68% of the participants were female, and the mean age was 46.3 years. Safety analyses were based on the actual treatment received (
Table 1), and efficacy analyses were based on the randomized intended treatment group.
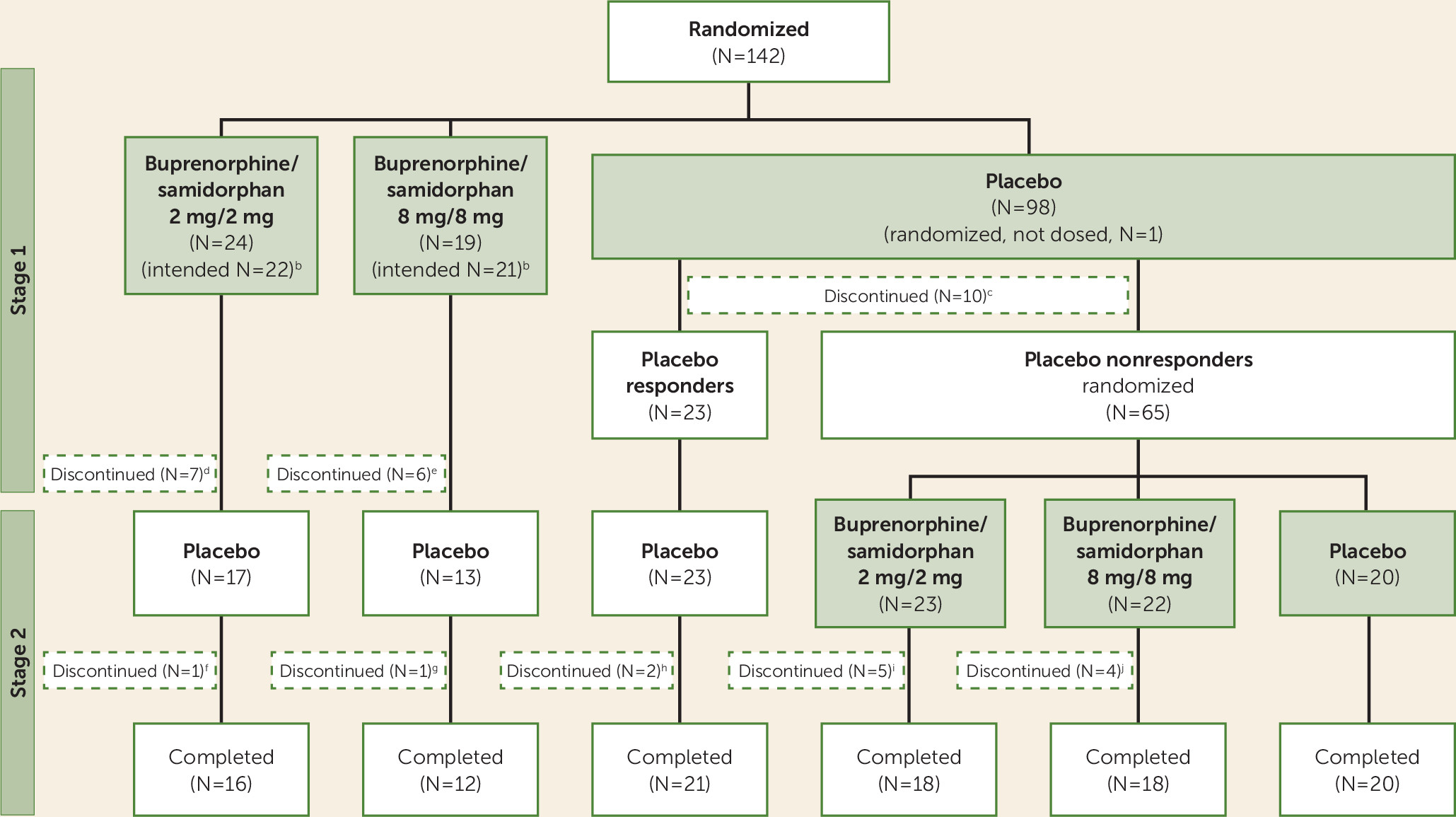
In the safety population, discontinuation rates during stage 1 were 10.2% (10/98 patients), 29.2% (7/24), and 31.6% (6/19) in the placebo, 2/2 dosage, and 8/8 dosage groups, respectively (
Figure 1). In stage 2, discontinuation rates were 5.5% (4/73), 21.7% (5/23), and 18.2% (4/22) in the placebo, 2/2 dosage, and 8/8 dosage groups, respectively.
Efficacy
Table 2 presents baseline and week-4 scores on the depression measures, as well as change from baseline, and
Table 3 presents placebo-adjusted change from baseline. As shown in
Table 3, there were significantly greater improvements in the 2/2 dosage group in placebo-adjusted least-squares mean differences on the HAM-D (−2.8, 95% CI=−5.1, −0.6; p=0.014), the MADRS (−4.9, 95% CI=−8.2, −1.6; p=0.004), and the CGI-S (−0.5, 95% CI=−0.9, −0.1; p=0.012) from baseline to the end of the 4-week treatment period in the overall study. There were smaller, nonsignificant changes in the 8/8 dosage group compared with the placebo group (on the HAM-D, −0.5, 95% CI=−2.8, 1.9; on the MADRS, −2.1, 95% CI=−5.6, 1.4; and on the CGI-S, −0.2, 95% CI=−0.6, 0.2). The overall effect size (Cohen’s d) for the 2/2 dosage group was 0.50 for the HAM-D and 0.54 for the MADRS. Sensitivity analysis using equal weighting for stages 1 and 2 rather than the 0.6/0.4 weighting resulted in a slightly larger treatment effect (see Table S2 in the online
data supplement).
In the stage-specific secondary efficacy analyses, a significant placebo-adjusted least-squares mean difference was also observed for the 2/2 dosage group on the HAM-D (−3.7, 95% CI=−6.9, −0.6, p=0.02), the MADRS (−6.7, 95% CI=−11.3, −2.0, p=0.005), and the CGI-S (−0.6, 95% CI=−1.2, −0.1, p=0.03) for stage 2 (
Table 3). No significant differences compared with placebo were observed for either of the buprenorphine/samidorphan treatment groups in stage 1 or for the 8/8 dosage group in either stage.
Response and Remission Rates
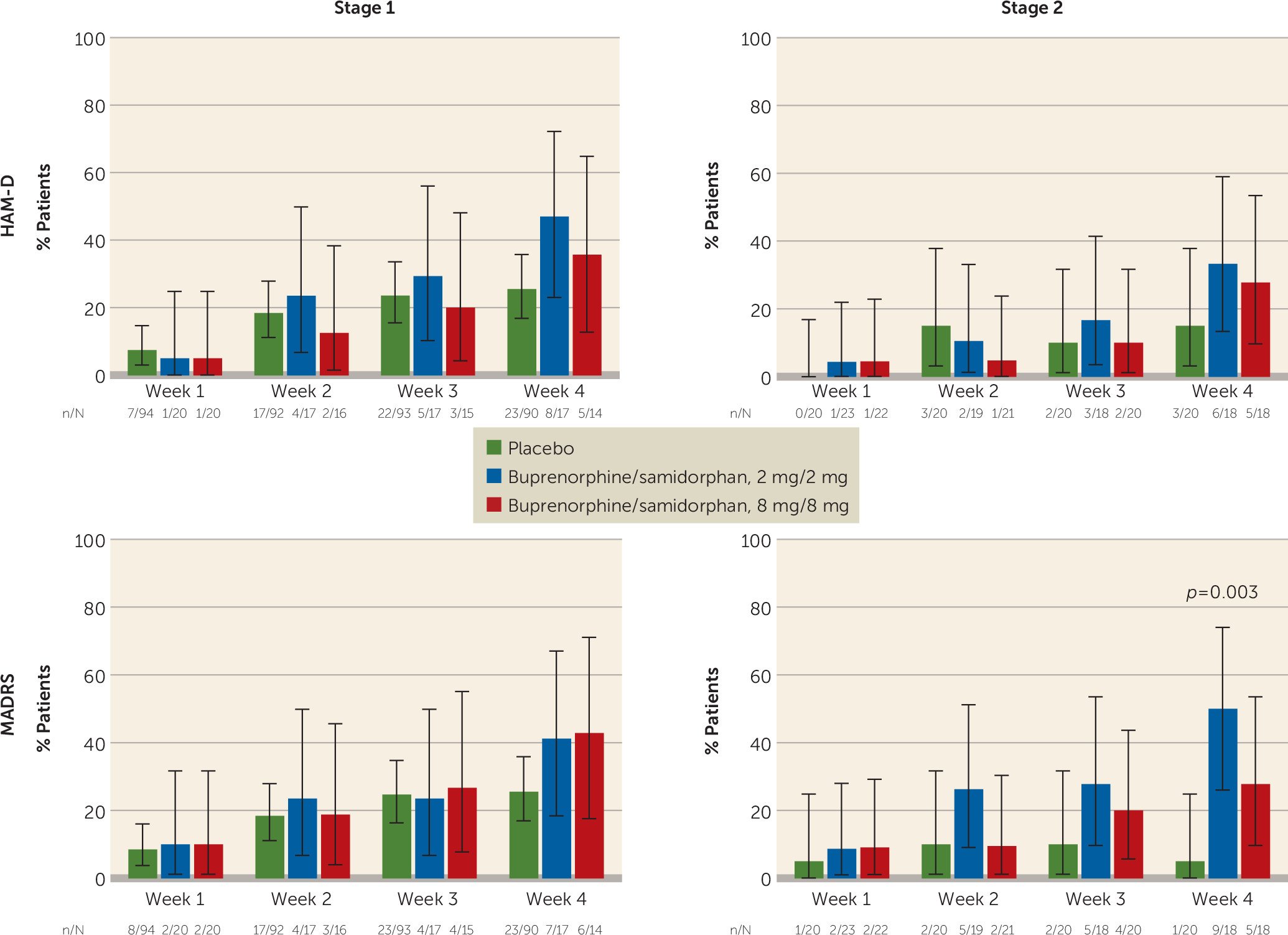
A greater proportion of patients in the active groups exhibited a treatment response according to the HAM-D (a reduction ≥50% in score from baseline to week 4) compared with placebo at week 4 in both stages (
Figure 2). Rates of response in stage 1 were 26%, 47%, and 36% for the placebo group, the 2/2 dosage group (p=0.086), and the 8/8 dosage group, respectively. In stage 2, HAM-D response rates were 15%, 33%, and 28% for the placebo, 2/2 dosage, and 8/8 dosage groups, respectively.
A greater proportion of participants in the active treatment groups had a treatment response according to the MADRS (a reduction ≥50% in score from baseline to week 4) compared with placebo at week 4 in both stages (
Figure 2). Rates of MADRS response in stage 1 were 26%, 41%, and 43% for the placebo, 2/2 dosage, and 8/8 dosage groups, respectively. In stage 2, MADRS response rates were 5%, 50% (p=0.003), and 28% for the placebo, 2/2 dosage, and 8/8 dosage groups, respectively. Rates of HAM-D and MADRS remission were similar to response rates (see Figure S1 in the online
data supplement).
Exploratory Measures
No statistically significant differences were observed for either of the buprenorphine/samidorphan dosage groups compared with the placebo group on the Inventory of Depressive Symptomatology–Self-Report, the Sheehan Disability Scale, or the SF-12.
Tolerability
The overall incidence of treatment-emergent adverse events was 85.8%, and the most common adverse events were gastrointestinal (52.5%) and neurological symptoms (46.8%). Common treatment-emergent adverse events occurring with buprenorphine/samidorphan were nausea, vomiting, dizziness, and headache (
Table 4).
Two patients (1.6%) in the placebo group and 17 (19.3%) in the buprenorphine/samidorphan groups discontinued because of treatment-emergent adverse events. The adverse events in one of the two placebo patients began while the patient was on placebo and ultimately led to discontinuation during stage 2 while the patient was taking the 8/8 dosage. The most common treatment-emergent adverse event leading to discontinuation was vomiting (4.3%). Similar proportions of patients in the 2/2 and 8/8 dosage groups had treatment-emergent adverse events leading to discontinuation. Most discontinuations in the buprenorphine/samidorphan groups occurred within the first 2 days of dosing. Serious treatment-emergent adverse events included an attempted suicide by multiple drug overdose (in the placebo group); intraocular melanoma (in the 2/2 dosage group); and acute opioid withdrawal (in the 2/2 dosage group), which occurred in a patient taking a prohibited opioid medication.
No evidence of opioid withdrawal was observed in assessments with the Clinical Opiate Withdrawal Scale during taper or follow-up periods. Generally neutral scores for drug liking were observed for all treatment groups. For the Addiction Research Center Inventory–Morphine Benzedrine Group, inconsistent results were observed in stages 1 and 2, with higher mean scores for the 2/2 dosage group compared with the other treatment groups in stage 1 but mean scores similar to those for the placebo group in stage 2 (see Table S3 in the online data supplement). The rate of emergence of suicidal ideation on the C-SSRS was low and similar in all treatment groups. No clinically relevant effects on laboratory test results, vital signs, or ECG were observed. Scores on item 11 of the HAM-D were consistent with a lack of change in suicidal ideation.
Urine drug screens to detect illicit substances were conducted at screening, baseline, and end of study. Among the 141 participants who entered the study and received study drug, one participant tested positive for benzodiazepines at screening but was negative at the baseline visit. A second participant tested positive for cocaine at the end of the study but was negative at the two previous visits. All other participants (N=139) were negative for all substances tested.
Discussion
Results of this trial demonstrate clinically meaningful antidepressant effects for the buprenorphine/samidorphan combination compared with placebo in patients with major depression and an insufficient response to SSRIs or SNRIs. The results expand the findings of a previous 1-week pilot trial in patients with treatment-resistant major depression (
22).
Significant differences for the 2/2 dosage group compared with the placebo group were noted on the HAM-D, the MADRS, and the CGI-S, and for response and remission on the MADRS. These results support the premise of the sequential parallel comparison design as a strategy to enhance signal detection in relatively smaller samples, and they are consistent with the finding that signal detection is enhanced in placebo nonresponders, as the effect size of buprenorphine/samidorphan was greater in stage 2 than in stage 1 (
24,
27).
The overall effect sizes for the 2/2 dosage were 0.50 for HAM-D and 0.54 for MADRS. The result compares favorably with results from a meta-analysis of 14 studies with atypical antipsychotics as adjunctive therapy for major depression, with reported effect sizes of 0.35 to 0.48 for individual drugs (
40).
The most common treatment-emergent adverse events observed were nausea, vomiting, and dizziness. These adverse events have been previously described with opioidergic agents. As most treatment-emergent adverse events occurred within the first 2 days of dosing, tolerability may be improved in future studies with slower titration of the 2/2 dosage. No evidence of opioid withdrawal was noted, and no consistent signal of abuse liability was observed.
Although there was evidence in both dosage groups of antidepressant activity, greater and statistically significant treatment effects were observed in the 2/2 dosage group. Although an inverse or U-shaped dose response is not uncommon for psychiatric medications, the specific reason for this observed dose response is uncertain. A potential explanation may relate to adverse events. Both the HAM-D and the MADRS contain questions that may be influenced by adverse events, including GI symptoms. Thus a higher incidence of vomiting and dizziness in the 8/8 dosage group may have contributed to the smaller reductions in HAM-D and MADRS scores. Future studies should explore the efficacy of buprenorphine/samidorphan dosages lower than 2 mg/2 mg.
Limitations of this study are the small sample size, the short duration of treatment, and the absence of long-term follow-up. Further clinical studies will also be needed to address definitively whether samidorphan successfully blocks the addictive potential of buprenorphine when these agents are administered in the 1:1 ratio. In addition, one might hypothesize that the sequential parallel comparison design results in attrition because of the longer period of time patients are receiving placebo. However, in augmentation depression trials, attrition is relatively modest as patients continue their current antidepressant treatment, and thus the use of the sequential parallel comparison design is justified.
These results support the hypothesis of a significant role of opioid dysregulation in major depression and the therapeutic potential of opioid modulation. Further investigation will be needed to understand the role and relative contribution of endogenous μ- and κ-opioid modulation in the observed antidepressant activity of the buprenorphine/samidorphan combination. The findings indicate that buprenorphine/samidorphan may be an important novel candidate for treatment of major depression, and support further confirmatory large-scale clinical trials.
Addendum: A recent press release by Alkermes (
http://phx.corporate-ir.net/phoenix.zhtml?c=92211&p=irol-corporateNewsArticle&ID=2131031) announced preliminary topline results from FORWARD-3 and FORWARD-4, the first two of three phase 3 efficacy studies from the comprehensive FORWARD pivotal program for ALKS 5461. FORWARD-4 tested two dosage levels of ALKS 5461 (2 mg/2 mg and 0.5 mg/0.5 mg) against placebo by using the sequential parallel comparison design described in this article. In that comparison, 385 patients entered the study. There was a clear trend toward efficacy on the primary endpoint with the 2 mg/2 mg dosage of ALKS 5461, and post hoc analyses achieved statistical significance for the entire 2 mg/2 mg dosage group on the MADRS endpoint. On the basis of these analyses, Alkermes believes that FORWARD-4 provides supportive evidence of the efficacy of ALKS 5461 in the treatment of major depressive disorder. FORWARD-3 tested ALKS 5461 (2 mg/2 mg) in comparison with placebo by using an alternate design. In that comparison, 429 patients entered the study. Placebo response was greater than that observed in FORWARD-4 and no treatment effect of ALKS 5461 was observed. Negative trials due to significant placebo effect are not uncommon in the study of major depressive disorder. FORWARD-5, the third pivotal efficacy study in the FORWARD program, is ongoing, testing two dosage levels of ALKS 5461 (2 mg/2 mg and 1 mg/1 mg) and using the sequential parallel comparison design. On the basis of information gained from FORWARD-3 and FORWARD-4, patient enrollment in FORWARD-5 will be increased and the statistical analysis plan will be updated.
Acknowledgments
The authors thank Mark S. Todtenkopf, Ph.D., for assistance in the preparation of the manuscript; the ALKS 5461 investigator study group; and Richard S. Perry, Pharm.D., for editorial assistance in the preparation of the manuscript, which was supported by Alkermes, Inc.