Major depression constitutes one of the most serious public health challenges worldwide (
1). Depression in older people, the most rapidly growing segment of the population, is associated with substantial morbidity and mortality from cardiovascular disease (
2) and suicide (
3), and it can be difficult to treat (
4). A substantial proportion of elderly patients with severe depression respond poorly to or cannot tolerate standard antidepressant treatments. For such patients, electroconvulsive therapy (ECT), widely acknowledged as the most effective acute antidepressant treatment, is an important option (
5). The use of ECT has been limited, however, by misperceptions about modern ECT technique and concerns about its cognitive effects (
6). Recent efforts to refine the technique have resulted in improved tolerability with largely preserved efficacy (
7). The use of right unilateral electrode placement with ultrabrief pulse width stimuli is a promising technical improvement that has gained widespread clinical use, with evidence for reduced cognitive side effects compared with conventional brief pulse ECT (
8,
9). Data supporting the use of right unilateral ultrabrief pulse ECT in the geriatric population are accumulating (
10) but still relatively modest.
Here, we present efficacy results for the initial phase of the multicenter Prolonging Remission in Depressed Elderly (PRIDE) study, funded by the National Institute of Mental Health (NIMH) and conducted by the Consortium for Research in ECT (CORE), in which 240 patients age 60 and over received right unilateral ultrabrief pulse ECT combined with venlafaxine.
Method
Design Overview
The PRIDE study was a multicenter randomized study of the relative efficacy, functional outcome, and tolerability of a novel strategy to enhance long-term outcomes in geriatric depression. The study design involved two phases: an acute ECT phase (phase 1) and a 6-month randomized maintenance phase (phase 2). In phase 1, patients received acute ECT three times per week as well as treatment with venlafaxine. In phase 2, patients who remitted in phase 1 were randomly assigned to receive either pharmacotherapy alone (venlafaxine plus lithium) or combined treatment with pharmacotherapy and continuation ECT enhanced with individualization of the ECT schedule.
The initial eight sites participating in PRIDE were the Icahn School of Medicine at Mount Sinai, New York (clinical coordinating center); Columbia University/New York State Psychiatric Institute, New York (clinical coordinating center); Medical University of South Carolina, Charleston (data management and statistical coordinating center); Zucker Hillside Hospital/Northwell Health System; University of Texas Southwestern Medical Center, Dallas; Mayo Clinic, Rochester, Minn.; Wake Forest University Medical Center, Winston-Salem, N.C.; and New York Presbyterian/Weill Cornell Medical Center, New York and White Plains. Columbia University/New York State Psychiatric Institute discontinued recruitment and study procedures in October 2010 and was replaced by Duke University School of Medicine, Durham, N.C., as a performance site and clinical coordinating center; Mayo Clinic discontinued recruitment in May 2012; Wake Forest University Medical Center discontinued recruitment in August 2012 and was replaced by Augusta University/Medical College of Georgia, Augusta. The trial was approved by the institutional review boards of the participating study sites.
Phase 1, reported here, was completed August 2014; phase 2 was completed in March 2015 and is reported separately (
11).
Patients
Participants were recruited from February 2009 through August 2014 from among patients who were referred for ECT at participating study sites. All patients provided written informed consent for research procedures and clinical ECT. Participants could be inpatients or outpatients and had to be at least 60 years old, have a DSM-IV diagnosis of unipolar major depressive episode, and have a score ≥21 on the 24-item Hamilton Depression Rating Scale (HAM-D) (
12). Exclusion criteria were diagnoses of bipolar disorder, schizoaffective disorder, dementia, or an intellectual disability; a diagnosis of substance abuse or dependence in the past 6 months; any active general medical or neurological condition that would have affected cognition or treatment response; contraindications to lithium or venlafaxine; and history of a failure to respond, in the current episode, to an adequate trial of venlafaxine plus lithium or to ECT.
Procedures
Medication washout and concomitant medications.
Psychotropic medications were discontinued within 1 week of starting phase 1. Rescue medication for agitation, anxiety, or insomnia was limited to lorazepam, up to 3 mg/day, or the equivalent.
ECT.
ECT procedures were standardized as follows: treatments were three times a week, with standard right unilateral electrode placement (d’Elia placement) (
13) using either a Somatics Thymatron System IV (Somatics, Lake Bluff, Ill.) with a pulse width of 0.25 ms and a current of 0.89 A or a MECTA spECTrum device (MECTA Corporation, Portland, Ore.) with a pulse width of 0.3 ms and a current of 0.8 A.
A dose titration procedure to determine seizure threshold was conducted at the initial treatment (see Tables S1 and S2 in the data supplement that accompanies the online edition of this article). Subsequent treatments were administered at six times the seizure threshold. The seizure adequacy criterion was a motor seizure ≥15 seconds, with restimulation if needed. If the HAM-D score demonstrated a decrease <25% from baseline by treatment 6, the stimulus dose was increased by 50%. If the HAM-D score demonstrated a decrease <25% from baseline by treatment 9, the stimulus dose was increased again by 50%.
Anesthesia procedures included administration of glycopyrrolate (0.2 mg i.v.) only at the dose titration session, induction with methohexital (∼1 mg/kg), muscle relaxation with succinylcholine (∼0.75 mg/kg), and ventilation with 100% oxygen throughout.
Medication.
Open-label venlafaxine was started 1–5 days prior to ECT or up to 2 days after the first treatment at an initial dosage of 37.5 mg/day, increased by 37.5 mg every 3 days or as tolerated, with a target dosage of 225 mg/day.
Assessments
Baseline assessments were obtained for eligible patients who provided informed consent. During study years 1 and 2, diagnosis was established using the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID-I) (
14). During study years 3–6, to minimize patient burden, the Mini International Neuropsychiatric Interview (
15) was used instead.
Depressive symptoms were assessed at baseline and three times per week thereafter using the HAM-D and the Clinical Global Impressions severity scale (CGI-S) (
16). Global cognitive function was measured using the Mini-Mental State Examination (MMSE) (
17) at baseline and at each treatment session (three times per week, prior to administration of ECT). Suicidal ideation was assessed using the Beck Scale for Suicide Ideation (
18). Baseline medical burden was assessed using the Cumulative Illness Rating Scale for Geriatrics (
19).
Ongoing consistency of HAM-D ratings across sites was addressed by having each rater score a new set of videotapes annually for comparison with the consensus ratings of the study co–principal investigator and the project coordinator. A more detailed description of the standardization of ratings is provided in the online data supplement.
Time to reorientation following ECT, measured after ECT treatments 1–3, was determined by assessing the patient’s ability to correctly answer five orientation questions (open eyes on command, name, age, birthday, day of the week). Patients were asked these items at 3, 5, 10, 15, and 20 minutes and were considered reoriented at the time point at which all five items were correctly answered. This method is modeled after that used in other studies (
7). Additional specific domains of neuropsychological functioning were assessed with a comprehensive neuropsychological test battery, the results of which will be reported elsewhere.
Safety measures assessed were serious adverse events and adverse events related to either ECT or venlafaxine, recorded according to Office for Human Research Protections guidelines. Safety measures were monitored externally by an NIMH data and safety monitoring board.
Primary Outcomes
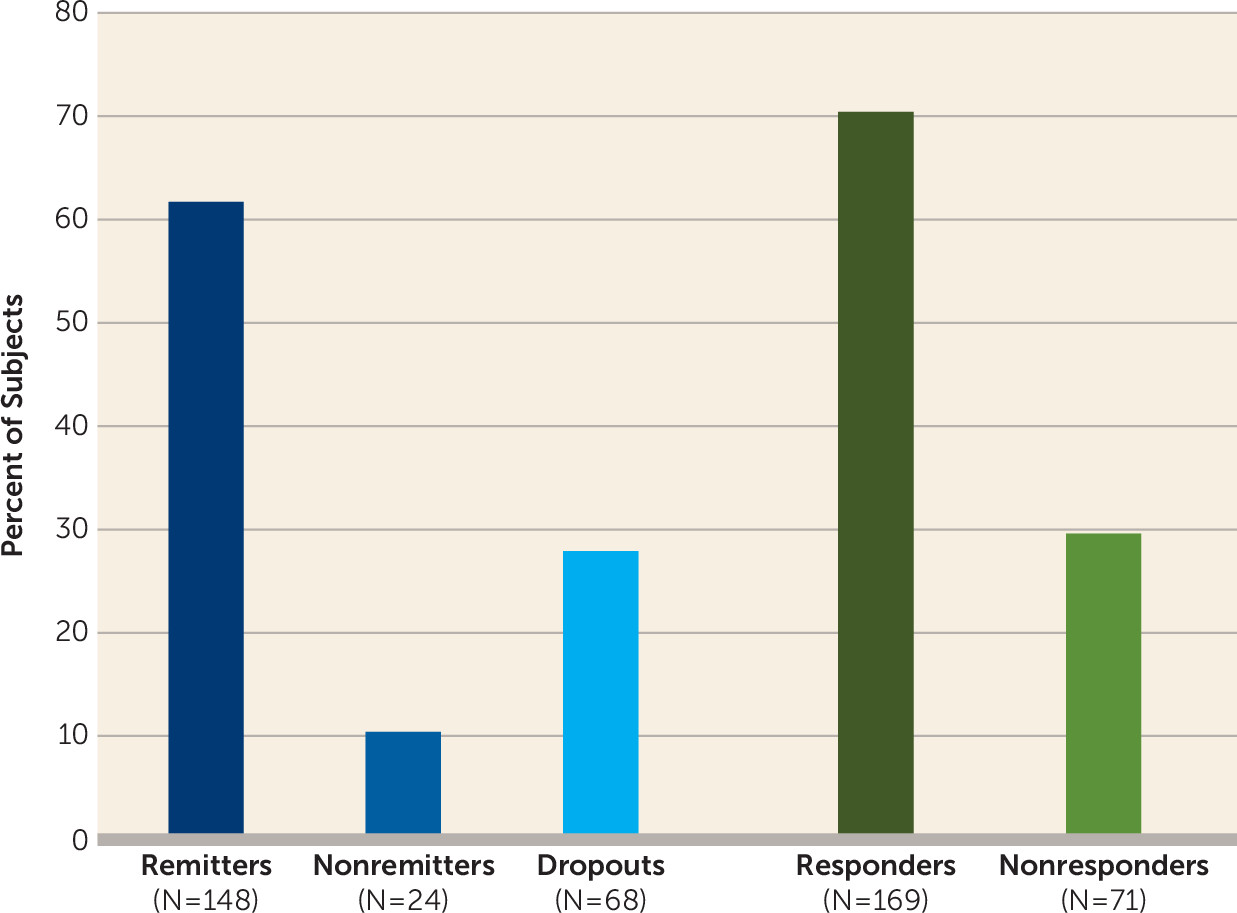
The primary outcomes in phase 1 were remission status and the longitudinal trajectory of HAM-D scores. Patients were classified as remitters if 1) they had a HAM-D score ≤10 on two consecutive ratings and 2) the HAM-D score did not increase >3 points on the second consecutive rating, or it remained ≤6. The minimum number of ECT treatments required for remission status was two, but there was no maximum. Patients were classified as nonremitters if they did not meet remission criteria, had at least 12 treatments, and reached a plateau, defined as no clinical improvement (<3-point decrease in HAM-D score after last two consecutive treatments). Patients were categorized as study noncompleters, or dropouts, if they did not meet remission or nonremission criteria and discontinued ECT before having received 12 treatments. There was no fixed time period required for completion of phase 1; patients could be declared remitters (and hence study completers) with as few as two ECT treatments, and nonremitters required at least 12 treatments to be considered study completers but could continue treatment if no plateau of improvement had been reached. Patients were considered study dropouts if they had received fewer than 12 ECT treatments before study exit and had not achieved remission.
Secondary efficacy outcomes in phase 1 were response status and speed of remission. Response was defined as a decrease of at least 50% from baseline in HAM-D score at the time of exit from phase 1. Speed of remission was determined by the number of ECT treatments required to achieve remission.
Statistical Analysis
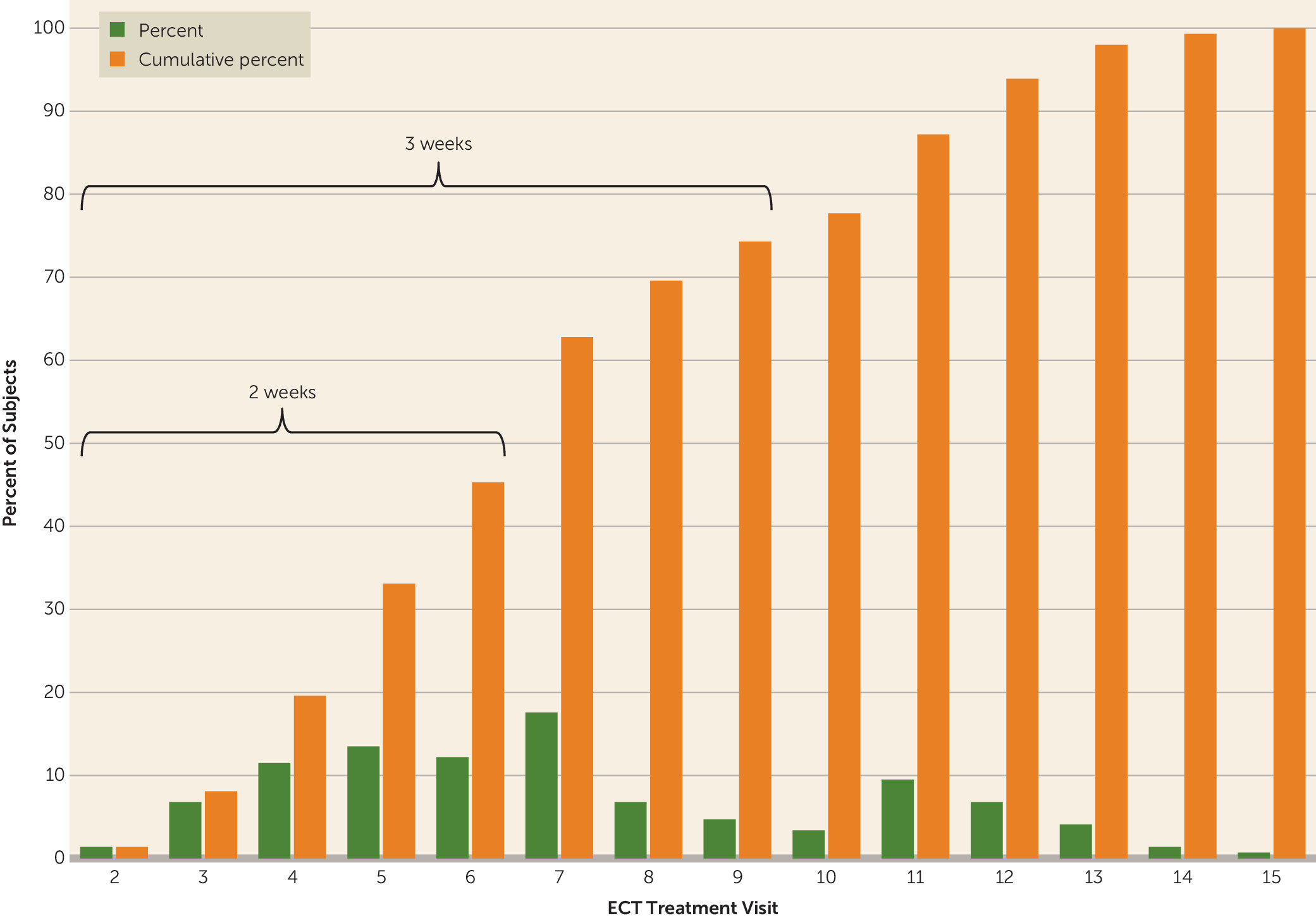
Frequency distributions for categorical variables and means with standard deviations for continuous variables were used to describe the phase 1 study sample. The proportions of remitters, nonremitters, and dropouts, with corresponding 95% confidence intervals, were determined at the point of exit from phase 1. There were no missing data for the primary three-level categorization (remitter, nonremitter, dropout), and no special methods for imputation were required. For some analyses, a dichotomous remission categorization was created by combining the nonremitter and dropout categories, an approach in which all study dropouts were considered to have had a negative (nonremitter) outcome. Similar analyses were carried out for response status, the secondary efficacy outcome. Speed of remission was estimated as the mean number of ECT treatments (with 95% confidence intervals) required to reach remission. In addition, we determined the percentage of remitters after specific numbers of ECT treatments—for example, percent who achieved remission after two treatments (the minimum required), within 1 week of treatment (three treatments), and within 2 weeks of treatment (six treatments).
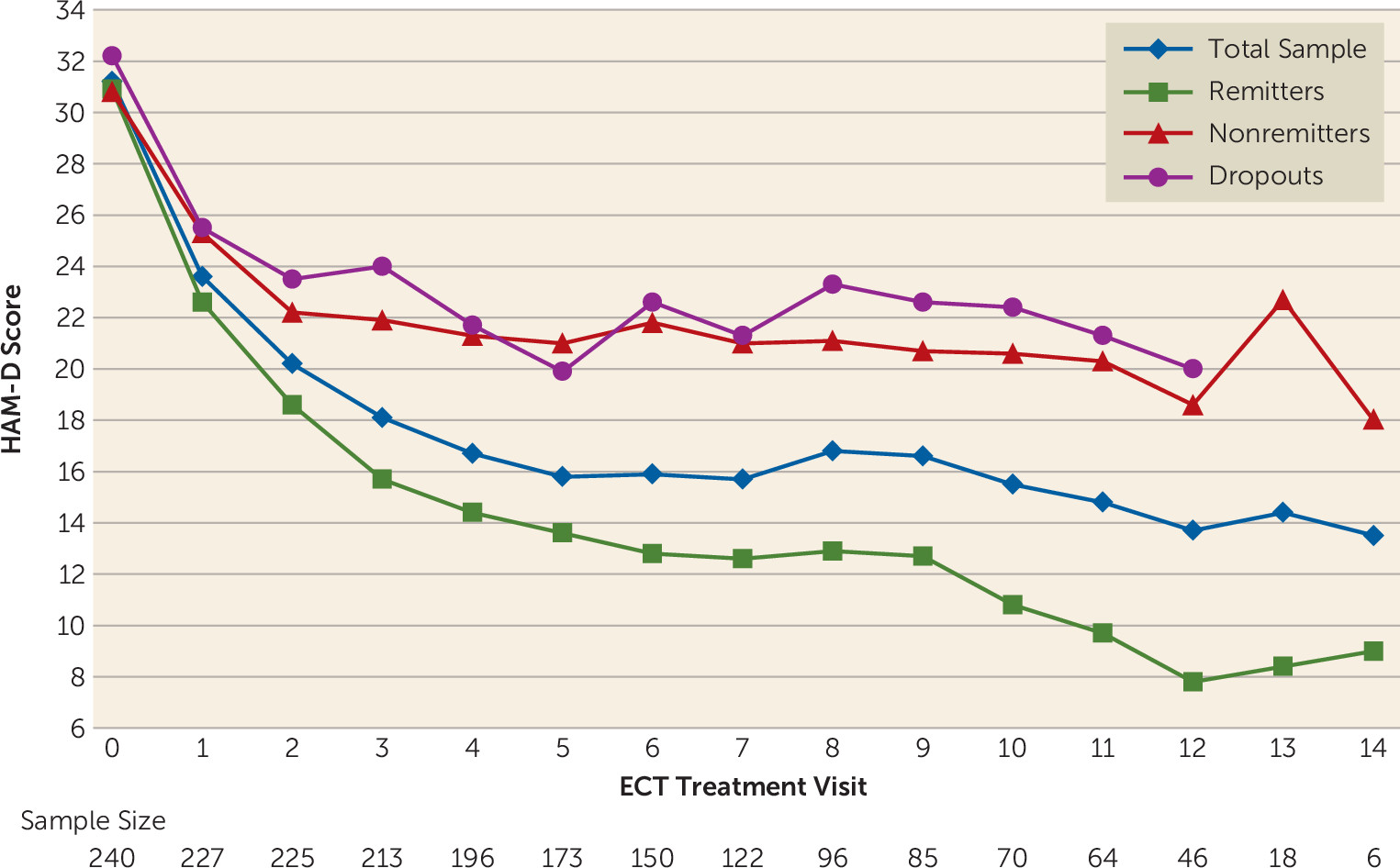
The observed means for HAM-D score at each ECT session were used to describe the trajectory of symptom severity through the course of phase 1 for the total sample and within outcome categories (remitters, nonremitters, dropouts). The magnitude of the mean change from baseline was estimated for the total sample and within outcome categories using 95% confidence intervals for difference in means; the significance of the change from baseline was assessed using the paired t test.
Analyses for MMSE scores were as described above for HAM-D scores. For time to reorientation, the proportions of patients falling into each time category (reoriented within 3 minutes, 5 minutes, and so on) were determined.
Predictors of outcome.
Given the similarity of characteristics for nonremitters and dropouts in most baseline and clinical variables (
Table 1), the dropout and nonremitter categories were combined for exploration of predictors of remission. Logistic regression was used to determine odds ratios and 95% confidence intervals. Considering all dropouts as nonremitters, a conservative approach, resulted in the combined category containing three patients with a last observed HAM-D score below 10 (but lacking two final HAM-D scores below 10, required for remitter status). A supportive analysis in which these three patients were omitted was carried out to test the sensitivity of results to the presence of these patients.
Discussion
These data add to the evidence base of the efficacy, safety, and tolerability of right unilateral ultrabrief pulse ECT in geriatric depression. Furthermore, they confirm a rapid speed of response to ECT in this cohort of severely depressed patients.
The antidepressant efficacy and speed of response observed in this study compare favorably with those reported in the few published studies of right unilateral ultrabrief pulse ECT. Sackeim et al. (
8) reported a remission rate of 73% for right unilateral ultrabrief pulse ECT in a cohort of 22 mixed-age adult patients. Mayur et al. (
20) found no efficacy difference between ultrabrief (N=17) and brief pulse (N=18) right unilateral ECT in a mixed-age population. Spaans et al. (
21) reported a remission rate of 41.4% in a cohort of 58 mixed-age patients who received right unilateral ultrabrief pulse ECT. More recently, Spaans et al. (
10) extracted 47 older patients (age 60 and over; mean age, 74 years) from the larger study cohort (brief pulse, N=21; ultrabrief pulse, N=26) and reported an overall remission rate of 64%. Loo et al. (
22), in a mixed-age cohort of 47 patients, reported a remission rate of 23.4% for right unilateral ultrabrief pulse ECT; however, in that study, end of treatment was determined on the basis of clinical judgment rather than a priori remission criteria.
The addition of venlafaxine was included as a feature of the PRIDE study design both to enhance the acute antidepressant effect of ECT (
23) (although a recent meta-analysis of ECT plus antidepressant compared with ECT alone failed to support this advantage [
24]) and to ensure that remitters in the acute phase (phase 1) would enter the randomized phase (phase 2) with the potential relapse prevention effect of antidepressant already initiated. Comparing the cohorts with the highest and lowest venlafaxine dosages in our sample revealed no difference in efficacy outcomes, suggesting that the largest part of the acute treatment effect was attributable to ECT.
In pooled data from our previous two CORE studies, bilateral electrode placement resulted in a remission rate of 69.7% in 244 patients age 60 and over (
25,
26). In the second CORE study, comparing the standard three electrode placements in ECT, brief pulse right unilateral ECT (pulse width, 1.0 ms) resulted in a remission rate of 70.4% in a cohort of 27 patients age 60 and over. The slightly lower remission rate of the present study relative to historical data using other forms of ECT is not unexpected and does not undermine right unilateral ultrabrief pulse ECT as a fully effective antidepressant treatment for a majority of geriatric depressed patients. Clinicians have the option of starting with right unilateral ultrabrief pulse ECT and crossing over patients who do not respond, as needed, to other ECT techniques (e.g., brief pulse right unilateral or bilateral electrode placement) (
27). Notably, the remission rates in this study are approximately twice those of antidepressant medications in similar populations (
28).
These data add to the relatively limited evidence base on the speed of response to right unilateral ultrabrief pulse ECT in geriatric depressed patients. Our mean of 7.3 ECT treatments (∼2.5 weeks) to reach remission compares favorably with medication (
29) and other forms of ECT in mixed-age populations (
30). In a systematic review, Tor et al. (
9) reported that the mean number of treatments for right unilateral ultrabrief pulse ECT was 9.6, whereas in our study only 26% of the patients required more than 10 treatments to achieve remission. Of note, we performed a midcourse dose increase in the event of slow or no response, which may have accelerated speed of response. Rapid response is clinically salient from the standpoint of minimizing illness burden, lowering treatment costs, minimizing the period of high suicide risk, and avoiding the reported risk of cognitive side effects with increasing numbers of treatments in older patients (
31).
The mean seizure threshold of 30.5 mC and the seizure elicitation rate of 84% at the first stimulus (25 mC, 5% of the device’s maximal output) demonstrate that this ultrabrief stimulus package is highly efficient at inducing seizures, even in a cohort of elderly patients, who would be expected on average to have relatively high seizure thresholds (
32). This allows for relatively low absolute stimulus doses to achieve dosing at six times seizure threshold, in turn minimizing the adverse cognitive effects of higher absolute stimulus charge (
33,
34).
Duration of disorientation after ECT sessions has been associated with the subsequent development of cognitive side effects, including retrograde amnesia (
35). Here we report relatively rapid reorientation, with close to half of patients being reoriented by 10 minutes.
The MMSE results reported here show little adverse effect of right unilateral ultrabrief pulse ECT on global cognitive functioning. The low rate of adverse and serious adverse events, including cognitive adverse effects, indicates that this form of ECT is well tolerated in geriatric depressed patients. The full results of the comprehensive neuropsychological test battery in phases 1 and 2 of the study will be reported elsewhere.
Strengths of this study include the moderately large sample size, standardized ECT and pharmacotherapy methods, and careful assessment of quality control. Shortcomings of phase 1 of the PRIDE study include the open trial design and the limited generalizability of the findings to the larger clinical ECT population, given that the study enrolled only geriatric patients with unipolar depression who had no serious medical or neurological comorbidities. Many of the most severely ill patients, who have a high likelihood of improvement with ECT, would have been ineligible for this study because they were too impaired to provide informed consent or would have been referred for alternative ECT techniques, including bilateral electrode placement (
36).