Schizophrenia is a serious, chronic mental illness with high heritability (64%−81%) (
1,
2). Important progress has been made in understanding the genetic basis of schizophrenia. Genome-wide association studies (GWAS) have identified more than 108 single-nucleotide polymorphisms (SNPs) that contribute to increased likelihood of schizophrenia (
3). However, the majority of SNPs contributing to schizophrenia liability fall short of genome-wide significance, and indices of polygenic risk incorporating larger proportions of SNPs have consistently demonstrated highly significant case-control differences (
3,
4).
Although common SNPs have weak individual effects (odds ratios, <1.2), several rare copy number variants (CNVs) have been identified that have a much stronger impact on risk (odds ratios, 2–57) (
5). Furthermore, an increased liability to schizophrenia has been associated with large deletions throughout the genome (
6,
7) and with an elevated overall CNV burden (
7–
9). For the specific deletions and duplications that confer risk for schizophrenia, only 1.4%−2.5% of individuals with schizophrenia carry one of them (
5). Risk for schizophrenia conferred by these CNVs is not deterministic, and many carriers do not develop schizophrenia. It is not known whether the additional factors affecting disease liability are environmental or reflect genetic variation within the CNV region or risk variants elsewhere in the genome.
Despite the strong effects from individual CNVs, the aggregate effect of common SNPs are at least an order of magnitude greater (
7,
10). Some overlap between GWAS and CNV findings for schizophrenia has been reported (
3,
7), and case subjects with associated CNVs have been shown to have elevated liability from common SNPs (
11). However, these two categories of genetic risk have generally been examined separately, and the relationship between them remains poorly understood.
In this study, we investigated the ways in which common SNPs and rare CNVs jointly contribute to the risk for schizophrenia. We tested a liability threshold model in which SNPs and CNVs act additively to confer disease risk. This model predicts that individuals with schizophrenia who have large-effect CNVs will, on average, have a smaller contribution from common SNPs. We also tested for interactions between common SNPs and specific CNVs. A second testable prediction from this model was that among control subjects, those with large-effect CNVs would typically have lower polygenic risk than control subjects without CNVs.
Method
Participants
We examined individuals from the Psychiatric Genomics Consortium schizophrenia study (
3) with available CNV data. Genome-wide genotype data from 33 independent European ancestry case-control samples were used (for further details, see Table S1 in the
online supplement). Each sample collection was approved by the relevant ethical review boards. All participants were at least 18 years old and provided written informed consent.
CNV Data
CNV data were derived from GWAS arrays and processed using a standardized pipeline by the Psychiatric Genomics Consortium CNV analysis group (for further details, see Marshall et al. [
12]). Briefly, multiple calling algorithms were applied to raw Illumina or Affymetrix intensity data from each individual. A consensus CNV call data set was generated by merging data at the sample level. After merging, arrays with excessive probe variance or guanine-cytosine bias were removed, as were samples with mismatches in sex, ancestry outliers, >7 Mb total CNV burden, or chromosomal aneuploidies. We removed samples with low-quality SNP genotyping and samples from individuals who were related to any other study subject. The final data set of rare, high-quality CNVs retained CNVs ≥20 kb, ≥10 probes, and frequency <0.01. CNVs that overlapped >50% with regions tagged as copy number polymorphic on any platform were excluded. Only autosomal chromosomes were used to facilitate comparability between sexes.
A total of 41,310 individuals met the above criteria (schizophrenia case subjects, N=21,088; control subjects, N=20,222).
Risk CNV Classes
Three categories of CNV risk were investigated. First, implicated loci were specific CNVs reported as genome-wide significant (for further details, see Table S2 in the online supplement). Carriers were defined as having ≥50% reciprocal overlap with reported CNVs (study subjects with overlap <50% were excluded from all analyses involving implicated loci). For NRXN1 deletions, each exon was considered separately. Six study subjects carried two implicated CNVs, and the CNV conferring the greatest risk was retained for analysis. Second, large CNV deletions (≥500 kb) anywhere in the genome were carried by 722 case subjects and 477 control subjects. Third, the total CNV burden for each study subject was examined.
Polygenic Risk Quantification
We generated risk profile scores by weighting each SNP by its log odds ratio in an independent set of GWAS results and applying these weights to SNPs in a second target data set. Summed across all SNPs, this yields a risk score for each study subject. These scores are a continuous and normally distributed measure of schizophrenia liability with highly significant differences between case and control subjects (
4,
3).
Risk profile scores were generated by conducting leave-one-out analyses (for further details, see the
online supplement and reference
3). Briefly, low frequency (<10%) and low-quality (imputation INFO <0.9) indels and SNPs in the extended major histocompatibility complex region (chr6:25–34 Mb) were excluded. We removed SNPs in high linkage disequilibrium and performed “clumping” (i.e., discarding variants within 500 kb and with r
2 ≥0.1 with a more significantly associated SNP). Polygenic scoring was performed using PLINK (
13) for multiple p-value thresholds (5×10
−8, 1×10
−6, 1×10
−4, 0.001, 0.01, 0.05, 0.1, 0.2, 0.5, and 1.0), multiplying the logistic regression weighting (i.e., the natural log of the odds ratio) of each variant by the imputation probability for the risk allele in each individual. The resulting values were summed over each individual to provide a whole genome risk profile score for further analysis.
Scores were then normalized to reduce between-cohort variation (for further details, see the supplemental data and Figure S1 in the online supplement). The information contained in the normalized scores was concentrated through principal component analysis (see the online supplement). The first principal component (polygenic risk score 1 [PRS1]) explains 69% of the total variability in the scores (see Figure S2A in the online supplement), was the only component associated with schizophrenia risk (odds ratio=2.40 [see Figure S2B in the online supplement]), and was used to index polygenic risk. This has the advantage of capturing the majority of polygenic risk in a single variable.
Statistical Models and Hypotheses
Intuitively, if the contributions of PRS1 level and CNVs to the risk of schizophrenia summed, we expected lower PSR1 levels among case subjects carrying CNVs compared with noncarrier case subjects. In the presence of CNV-mediated risk, a lower PSR1 would be sufficient to place case subjects over the threshold for schizophrenia. A similar argument holds for control subjects, whereby control subjects with CNVs and high PRS1 levels would be underrepresented compared with control subjects without CNVs.
More formally stated, since PRS1 and CNV status are both positively associated with the risk of schizophrenia, we hypothesized that an additive liability model with an increasing link function would predict lower PRS1 values for individuals strongly influenced by the presence of a previously associated CNV, a large deletion, or high total CNV burden. Because this prediction holds for both case and control subjects separately, we were able to test the following core hypotheses: 1) for schizophrenia case subjects, the mean PRS1 among CNV carriers and individuals with higher total CNV burden would be lower than that for noncarriers and individuals with lower total CNV burden, and 2) for control subjects, the mean PRS1 among CNV carriers and individuals with higher total CNV burden would be lower than that for noncarriers and individuals with lower total CNV burden.
Both hypotheses were tested with respect to three CNV measures, although power was limited for testing in the control group because of the rarity of schizophrenia-associated CNV variants. For specific CNVs and large deletions, we tested differences in mean PRS1 levels between carriers and noncarriers using a two-sided Welch t test. For total CNV burden, we regressed PRS1 levels on total CNV burden and used a two-sided Wald t test to test for negative slopes among case and control subjects.
On the basis of our findings, we also fitted logistic regression models with schizophrenia status as outcome, PRS1 and CNV status as predictors, and adjustment for site, sex, CNV quality, and five ancestry principal components. These models were fitted separately for carriers and noncarriers of specific CNVs. By comparing a series of nested models via likelihood ratio tests and measures of model fit, we could quantify the contribution of PRS1 and CNV both individually and jointly as well as test for nonadditive effects in modeling schizophrenia risk (for further details, see the online supplement). For models with statistically significant nonadditive effects, we report predicted odds ratios to demonstrate the pattern of nonadditivity.
The threshold for significant test results was set to 5%. No multiple testing correction was applied because the statistical tests were not independent.
Results
The numbers of case and control subjects carrying each type of risk CNV are presented in
Table 1. Among case subjects, the mean polygenic risk score for CNV carriers was significantly lower than that for noncarriers (carrier group: PRS1=0.70; noncarrier group: PRS1=0.97; p=0.03) (
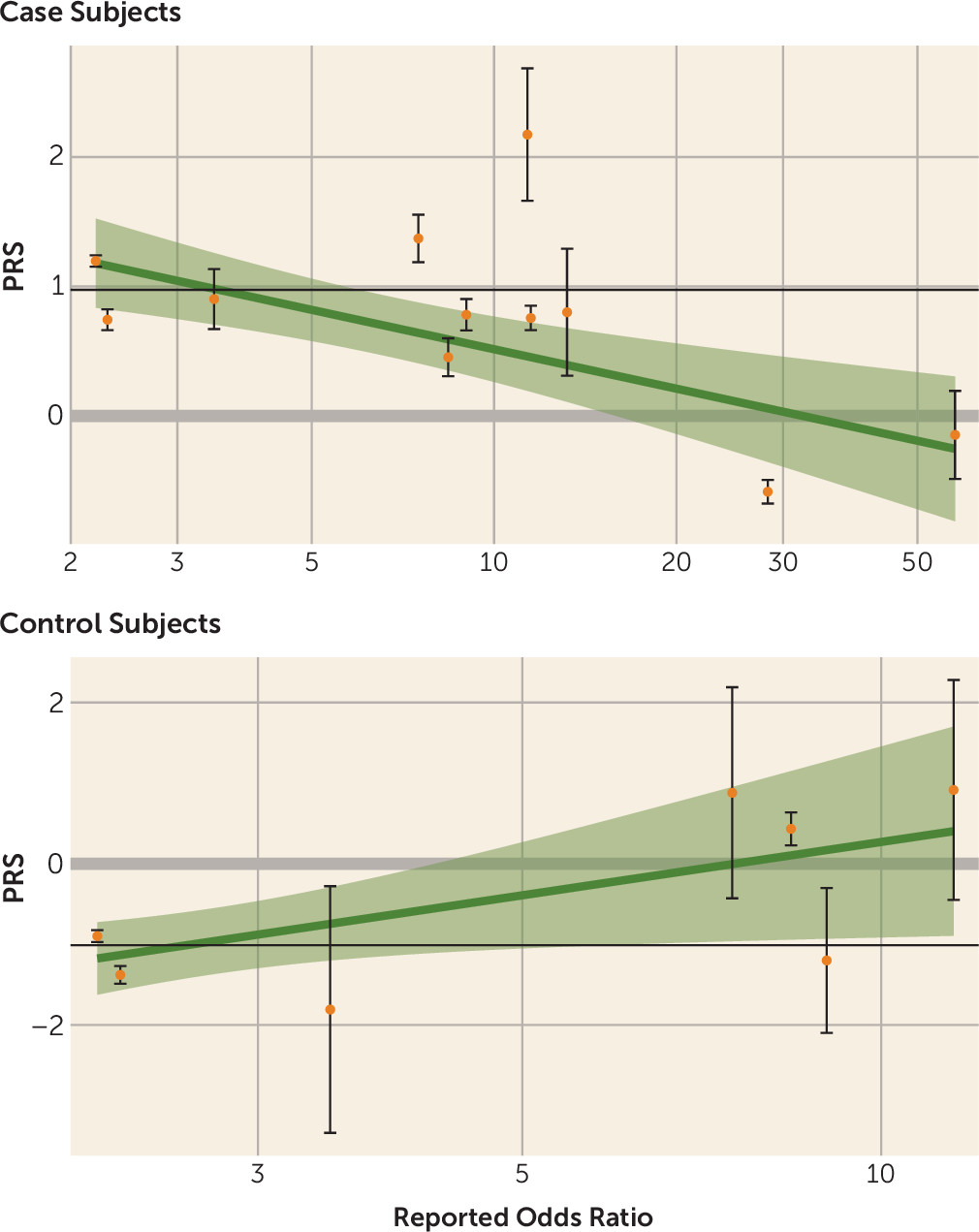
Table 2). This relationship was stronger with increasing risk from the specific CNVs. When the CNVs were divided into three groups based on the odds ratio of their association with schizophrenia (odds ratio ranges, <5, 5–15, and >15), only the carriers of CNVs with odds ratios >15 had a significantly lower PRS1 score than the noncarriers (
Table 2). The relationship for individual CNVs is illustrated in
Figure 1. On average, the mean PRS1 value for carriers of an individual CNV decreased with the effect size (odds ratio) of the CNV. For example, we found that whereas case subjects with 15q11.2 deletions (odds ratio=2.2) (
5) had a mean PRS1 close to what we observed for noncarrier case subjects, case subjects with 22q11.2 or 3q29 deletions (odds ratios, 28.3-∞ and 57.7, respectively) (
5) had much lower PRS1 scores (
Figure 1) (see also Table S3 in the
online supplement).
For control subjects, the relationship was unexpectedly reversed, because carriers of CNVs with larger effect sizes had significantly higher mean risk scores (
Table 2). Statistical significance and effect size were less clearly tied to reported odds ratios for control subjects than for case subjects.
Case subjects with large-deletion CNVs had reduced PRS1 compared with noncarrier case subjects (PRS1=0.77 compared with PRS1=0.98, p=0.02). However, upon removal of case subjects carrying CNVs previously implicated to increase the risk for schizophrenia, the results became nonsignificant (PRS1=0.89 compared with PRS1=0.98, p=0.43). No statistically significant differences were observed for control subjects (
Table 3).
Increasing the total CNV burden was associated with significantly decreased PRS1 among case subjects (reduction of mean PRS1 by 1.05 for each 10-kb extra CNV, p=0.0024) but not control subjects (increased mean PRS1 by 0.19, p=0.65) (
Table 4). When the CNVs previously implicated in schizophrenia risk were removed, the burden of the remaining CNVs was not significantly associated with PRS1 (p=0.08).
Model-Fitting Results
For noncarriers of CNVs previously shown to be associated with schizophrenia, PRS1, large deletions, and total CNVs were individually significant (models 1–3) (for further details, see Table S4 in the online supplement). Both large deletions and total burden add significantly in an additive manner to PRS (models 4 and 5), with no indication of significant interactions (models 6 and 7).
For carriers of these previously associated CNVs, the genetic risk score and the log(odds ratio) of the specific CNV as well as other large deletions had significant predictive power (models 1–3) but not total burden (model 4) (for further details, see Table S4 in the online supplement). Adding the log(odds ratio) to PRS1 improved the model significantly (model 5), and there was a significant interaction (that is, a nonadditive effect) between PRS1 and log(odds ratio) (model 7). Similarly, adding large deletions to PRS1 conferred significant improvement (model 6), and there was a significant interaction between them (model 8). The interaction parameter was negative in both models 7 and 8, meaning that increasing PRS1 levels had less impact on the risk of schizophrenia in carriers of a specific CNV compared with noncarriers. However, once the PRS, the CNV effect size, and their interaction were properly accounted for, large deletions conferred no improvement (model 9).
The interaction between PRS1 and effect sizes for individual CNVs (model 7) is summarized in
Table 5 (see also Table S4 in the
online supplement). We report the predicted odds ratio for schizophrenia associated with an increase of PRS1 by one unit, sorted by the reported effect size of the individual CNVs (smallest to largest) (see Table S2 in the
online supplement). We also included the corresponding predicted odds ratio for noncarriers (based on model 1) as a reference (see Table S4 in the
online supplement). Only for carriers of the three CNVs with the lowest reported effect sizes (15q11.2 deletions, 16p13.11 duplications, and 1q21.1 duplications) did we observe statistically significant evidence that an increase in the PRS actually increased the risk of schizophrenia (all variants, p≤5.5×10
−9). The associated predicted odds ratios (1.41–1.56) were slightly in excess of the predicted odds ratio for noncarriers (1.40), although not statistically significantly (all variants, p>0.11).
Crucially, the results for the interaction model for specific loci are in line with the results of testing the original two hypotheses for this CNV category: because of the smaller contribution of PRS1 to the total risk of schizophrenia among carriers of medium-risk to high-risk loci, the model implies that case subjects who carry a specific CNV with higher reported risk would have a lower mean PRS1 compared with case subjects who do not.
Discussion
The goal of this study was to clarify how aggregate measures of common risk SNPs and rare CNVs jointly contribute to the risk for schizophrenia. Five results were noteworthy. First, as predicted by an additive model, aggregate affected carriers of previously identified CNVs for schizophrenia had significantly lower PRSs than affected noncarriers. Second, when we subdivided these CNVs by effect size, the significant reduction of PRS was only seen for the CNVs with the strongest effect on schizophrenia risk. Third, while all case subjects with large deletions cumulatively had a significantly lowered PRS, when we eliminated case subjects with previously implicated CNVs, this effect disappeared. Fourth, the total CNV burden among case subjects was significantly and inversely related to the PRS. The effect was entirely the result of deletions, whereas duplications had no effect. Furthermore, as with the large deletions, when we removed case subjects with known CNVs, this relationship disappeared. Finally, our formal modeling revealed an additive relationship between the PRS and either total CNV burden or large CNVs, meaning that the risk for schizophrenia was well captured by simply taking the sum of these two kinds of genetic risks. However, when we examined individual CNVs, we found a more complex relationship: increasing PRS levels had less influence on the risk of schizophrenia among carriers of large-effect CNVs compared with carriers of small- to moderate-effect CNVs.
Our results are congruent with a previous report that specific previously associated CNVs require a genomic context of liability to result in schizophrenia (
11), supporting the conclusion that these loci do not represent fully penetrant Mendelian forms of illness. Our analyses of 41,321 individuals included the 11,428 study subjects previously reported. And in addition to establishing more conclusively that carriers of schizophrenia-associated CNVs generally require elevated genomic risk, we tested and confirmed that these forms of genetic risk act in an interactive manner. Additionally, we were able in this study to evaluate individual CNV-PRS relationships, yielding important results for carriers as well as researchers generating disease models involving these CNVs. Furthermore, this study tested relationships between polygenic risk and other well-replicated categories of CNVs causing risk, large deletions throughout the genome, and total CNV burden.
For large-deletion carriers and high total CNV burden, the lower PRS1 observed in case subjects was primarily driven by carriers of the implicated CNVs and was not mirrored in the results for healthy control subjects. This may have been due to the genomic locations of these CNVs in the two study groups, because the case subjects more often carried CNVs intersecting regions of genomic risk for schizophrenia. Furthermore, schizophrenia case subjects comprised only a small portion of one tail of the liability distribution. Therefore, a small elevation in risk from CNVs in control subjects was not likely to have a detectable effect at most points along the liability curve. These results also suggest that the specific CNVs conferring substantial risk for schizophrenia have likely all been identified.
The only results inconsistent with our original hypotheses was the observation of greater polygenic loading in control subjects with any previously implicated CNV compared with noncarrier control subjects. Data on the ages for the control subjects were not available; many of them may have been young and still within the age at risk. Also, not all samples used screened control subjects; some could have had schizophrenia or developed schizophrenia later, contributing to the observed results. Because both CNV and polygenic risk could drive behavioral characteristics in a similar direction, assortative mating could produce coaggregation, but not specifically in control subjects. This would be more likely in carriers of the low-effect-size CNVs, which are more often inherited.
Different patterns of results may exist across diseases, indicating different genetic architectures. However, one investigation of children with attention deficit hyperactivity disorder (ADHD) found a similar pattern (
15). ADHD case subjects with large deletions (>500 kb) (N=60) had lower ADHD PRSs compared with other affected children (N=421) Additional studies of other complex genetic diseases could offer a broader understanding of the range of genetic architectures underlying neuropsychiatric disorders.
Six potential limitations should be considered in the interpretation of our results. First, even in this study, involving, to our knowledge, the largest number of schizophrenia case subjects to date with CNV data, power to detect effects in the carriers of the specific risk CNVs was limited by their rarity. Second, rare single-nucleotide variants identified through DNA sequencing comprise a third class of genetic variation contributing to schizophrenia risk (
10). Such data were not available for the samples analyzed and therefore were not incorporated into these analyses. Third, copy number polymorphisms with >1% frequency, which are rarely investigated, were not examined. Fourth, although we tested interactions between CNVs and aggregate SNPs, epistatic interactions may exist between specific CNVs and specific risk SNPs that are beyond the scope of this study. Fifth, SNPs included in the polygenic scoring falling within the previously associated CNV regions could have biased analyses of interactions. Because only 399 of 102,636 SNPs fell in these loci, this is unlikely to have influenced our results. Finally, carriers of some implicated CNVs (particularly 22q11.2) who do not develop schizophrenia are unlikely to be recruited as control subjects, because of medical problems or intellectual impairment. This may produce inflated estimates of schizophrenia risk and complicates interpretation of the relationship between CNV effect sizes and genomic risk. We therefore conservatively used the lower-bound estimate of the 22q11.2 effect size. It is noteworthy that in a previous sample of 329 carriers of this deletion, those who developed schizophrenia were significantly more likely to have additional CNVs affecting genes relevant to this disorder (
16).