Major depressive disorder is one of the most widespread psychiatric illnesses, affecting an estimated 300 million people worldwide (
1) and carrying enormous personal and socioeconomic consequences (
2). Neuroimaging studies demonstrate reduced volume of the prefrontal cortex (PFC) and hippocampus (
3,
4), where neuronal atrophy and glial loss have been reported in postmortem brains from depressed subjects and rodent chronic stress models (
5,
6). Although the mechanisms underlying the pathophysiology and treatment of depression are still unknown, there is mounting evidence indicating a role of growth factors, notably brain-derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF). Chronic stress and depression decrease the expression and function of BDNF and/or VEGF in the PFC and hippocampus (
7–
11); VEGF is also decreased in the CSF of patients who have attempted suicide (
12). In contrast, chronic, but not acute, treatment with typical antidepressants, notably monoamine reuptake inhibitors, increases BDNF and VEGF expression (
7,
13–
17), and blockade of BDNF or VEGF signaling attenuates the antidepressant effects of these treatments (
13,
17,
18). These findings support a neurotrophic hypothesis of depression that reduced neurotrophic factor levels are strongly linked with the structural alterations caused by chronic stress and depression and, conversely, that antidepressants act at least in part by producing the opposite effects via induction of BDNF and VEGF expression.
In contrast to the delayed response of weeks to months for the action of typical antidepressants, a single subanesthetic dose of ketamine, an
N-methyl-
d-aspartate receptor antagonist, produces rapid (within hours) and sustained (up to 1 week) antidepressant actions even in patients with treatment-resistant depression (
5,
19). Preclinical studies have reported that low-dose ketamine produces rapid, sustained antidepressant behavioral responses and increases the number and function of synapses in medial PFC (mPFC) pyramidal neurons, reversing the synaptic loss caused by chronic stress (
20,
21); these effects are dependent on the processing and activity-dependent release of BDNF (
22,
23). Together, these findings indicate that the neurotrophic responses are associated with the behavioral actions of ketamine, but it remains unclear whether VEGF signaling is also involved. VEGF is a pleiotropic growth factor expressed by neurons and glia, as well as vascular endothelial cells (
18), and has potent neurotrophic activity (
24). Here, we examined the role of neuronal VEGF signaling in the behavioral and neurotrophic effects of ketamine.
Methods
Animals
Male wild-type CaMKIIα-Cre (
25), Flk-1
flox/flox (
26), VEGF
flox/flox (
27), CaMKIIα-Cre:Flk-1
flox/flox (hereafter, Flk-1
neuron−/−), and CaMKIIα-Cre:VEGF
flox/flox (hereafter, VEGF
neuron−/−) mice and Sprague-Dawley rats were used. Animal use and procedures were in accordance with National Institutes of Health guidelines and were approved by the Yale University Animal Care and Use Committee.
Surgery and Drug Infusion
Guide cannulas were implanted bilaterally into the mPFC (rats, 3.0 mm rostral, ±1.0 mm lateral, 4.0 mm ventral to the bregma; mice, 1.8 mm rostral, ±0.4 mm lateral, 2.5 mm ventral to the bregma) or dorsal striatum (rats, 1.6 mm rostral, ±1.9 mm lateral, 5.0 mm ventral to the bregma). Rats were bilaterally infused with a VEGF neutralizing antibody or rat recombinant VEGF164 using an injector that protruded 0.5 mm beyond the tip of the guide cannula (0.5 μL/side; 0.25 μL/min). Mice were bilaterally infused with ketamine or mouse recombinant VEGF164 using an injector that protruded 0.3 mm beyond the tip of the guide cannula (0.2 μL/side; 0.2 μL/min).
AAV2shFlk-1 Preparation and Intra-mPFC Viral Infusion
Sense and antisense oligonucleotides encoding the Flk-1 short hairpin RNA (shFlk-1) were ligated into a plasmid (pshFlk-1) designed to restrict shRNA expression to cells that express Cre recombinase, as has been previously reported (
28). This plasmid allows U6 promoter-dependent shFlk-1 expression only when cytomegalovirus promoter-enhanced green fluorescent protein (CMV-EGFP) reporter/stop cassette is eliminated by Cre-dependent recombination.
Sense: 5′-TTAAACCGGGATGTGAAACCCTTTCTTCAAGAGAGAAAGGGTTTCACATCCCGGTTTATTTTTTC-3′
Antisense: 5′-TCGAGAAAAAATAAACCGGGATGTGAAACCCTTTCTCTCTTGAAGAAAGGGTTTCACATCCCGGTTTAA-3′
To package into AAV2shFlk-1, HEK293 cells were transfected with pHelper, pAAV2-RC, and pshFlk-1 constructs using Lipofectamine 2000 (Life Technologies, Carlsbad, Calif.). Forty-eight hours after transfection, AAV2shFlk-1 particles were purified, resuspended, and stored at −80°C. Mice were infused with AAV2shFlk-1 bilaterally into the mPFC (2.0 mm rostral, ±0.3 mm lateral, 2.8 mm ventral to the bregma; 1 μL; 0.15 μL/min).
Behavioral Testing
A forced swim test, novelty-suppressed feeding test, and female urine sniffing test were performed, and these have been described elsewhere (
17,
22,
28–
31). In the forced swim test, 24 hours after a preswim, each animal was placed in a swim cylinder for 10 minutes and videotaped. The immobility time was scored during minutes 2 through 6. In the novelty-suppressed feeding test, animals were food-deprived overnight and placed in an open field with a small amount of food in the center. The latency to feed was measured. In the female urine sniffing test, animals were exposed to a cotton-tipped applicator infused with fresh urine from females of the same strain for 5 minutes. The time spent sniffing the cotton-tipped applicator was measured. In the tail suspension test, each mouse was suspended by its tail for 6 minutes and videotaped. The immobility time was measured during the last 4 minutes. In the locomotor activity test, each animal was placed in a testing chamber for 20 minutes. The total distance traveled (mice) or the number of beam breaks (rats) was measured.
Immunohistochemistry
After 3,3′-diaminobenzidine staining, the number of c-Fos-positive cells was counted in the regions near the infusion sites in the mPFC from two coronal sections (40 μm) in each rat.
Immunofluorescence
Free-floating sections (30 μm) were incubated with the following primary antibodies: rabbit anti-CaMKII, mouse anti-CaMKII, goat antimouse Flk-1, mouse antiglucose transporter 1, and rabbit anti-VEGF. They were then incubated with secondary antibodies. To quantify knockdown, the number of Flk-1-positive cells were counted and calculated as the percentage of total DsRed+ cells.
Golgi Staining and Spine Density Analysis
Golgi staining was performed using the FD Rapid GolgiStain kit (FD NeuroTechnologies, Columbia, Md.) according to the manufacturer’s instructions. Spine density on the primary and secondary dendritic branches of the apical tuft of layer V pyramidal neurons in the infralimbic and prelimbic regions of the mPFC was analyzed using Neurolucida 10 (MBF Bioscience, Williston, Vt.).
Sholl Analysis
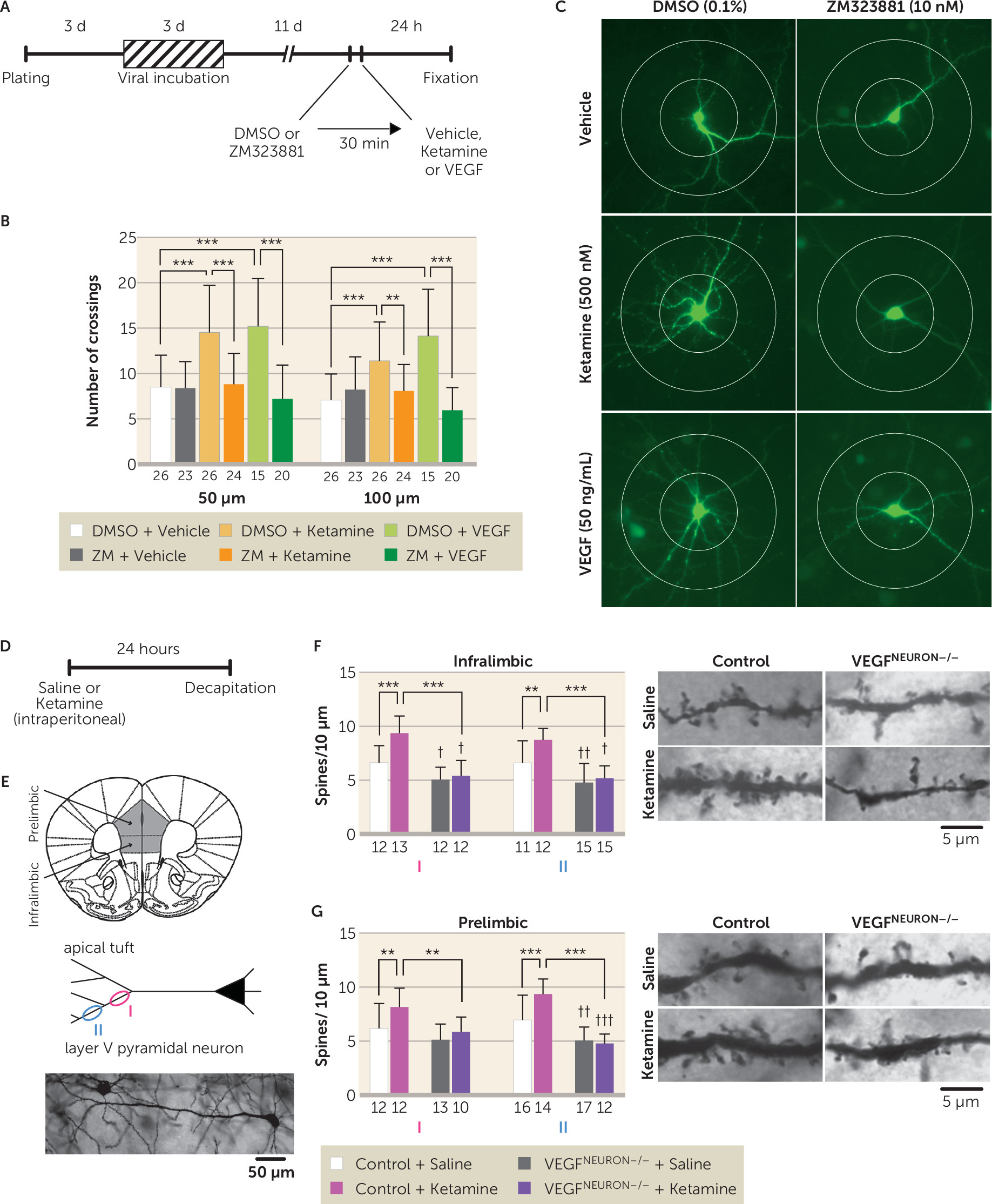
Dendritic complexity in primary cortical neurons was analyzed as described elsewhere (
32). Cortical neurons were dissected from E18 rat embryos, incubated with AAV2-EGFP for 72 hours, and treated with 500 nM of ketamine or 50 ng/mL of VEGF with or without the Flk-1 inhibitor ZM323881. After 24 hours of incubation, the number of dendritic crossings 50 μm and 100 μm from the soma was measured.
Statistics
Data are expressed as means and standard deviations. Data were analyzed by unpaired t test, one-way analysis of variance (ANOVA), or two-way ANOVA followed by Fisher’s least significant difference test using GraphPad Prism 6 (GraphPad Software). Differences with p<0.05 were considered statistically significant.
Detailed methods are available in the online supplement.
Discussion
The results demonstrate that neuronal VEGF–Flk-1 signaling in the mPFC plays a pivotal role in the antidepressant-like behavioral and neurotrophic actions of ketamine. We also found that a single intra-mPFC infusion of ketamine or VEGF
164 mimics the behavioral effects of systemic ketamine in a neuronal Flk-1–dependent manner. These findings indicate that neuronal VEGF–Flk-1 signaling in the mPFC is necessary and sufficient for the rapid and sustained antidepressant-like behavioral actions of ketamine. Alternative splicing of the VEGF gene results in various isoforms of different amino acid lengths, with VEGF
120 and VEGF
164 the most abundant (about 20% and 75%, respectively) in adult mouse brain (
33). Both VEGF
120 and VEGF
164 bind two high-affinity tyrosine kinase receptors, Flt-1 (VEGFR1) and Flk-1 (VEGFR2), but only VEGF
164 binds to the coreceptors neuropilin-1 and -2 (
34). Our results demonstrate that Flk-1 is required for the behavioral actions of ketamine and VEGF
164, although we cannot rule out the contribution of Flt-1 and neuropilin-1 and -2.
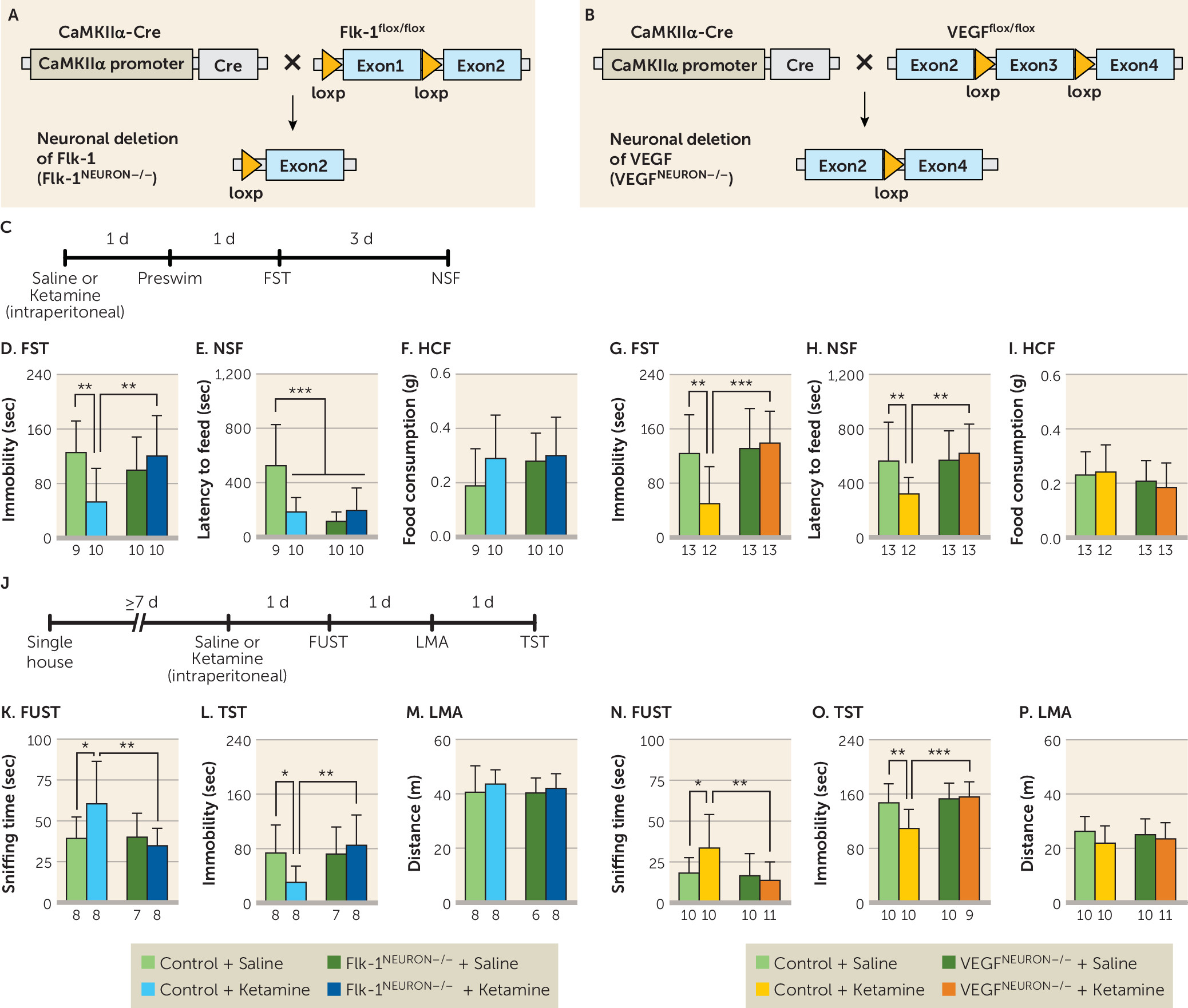
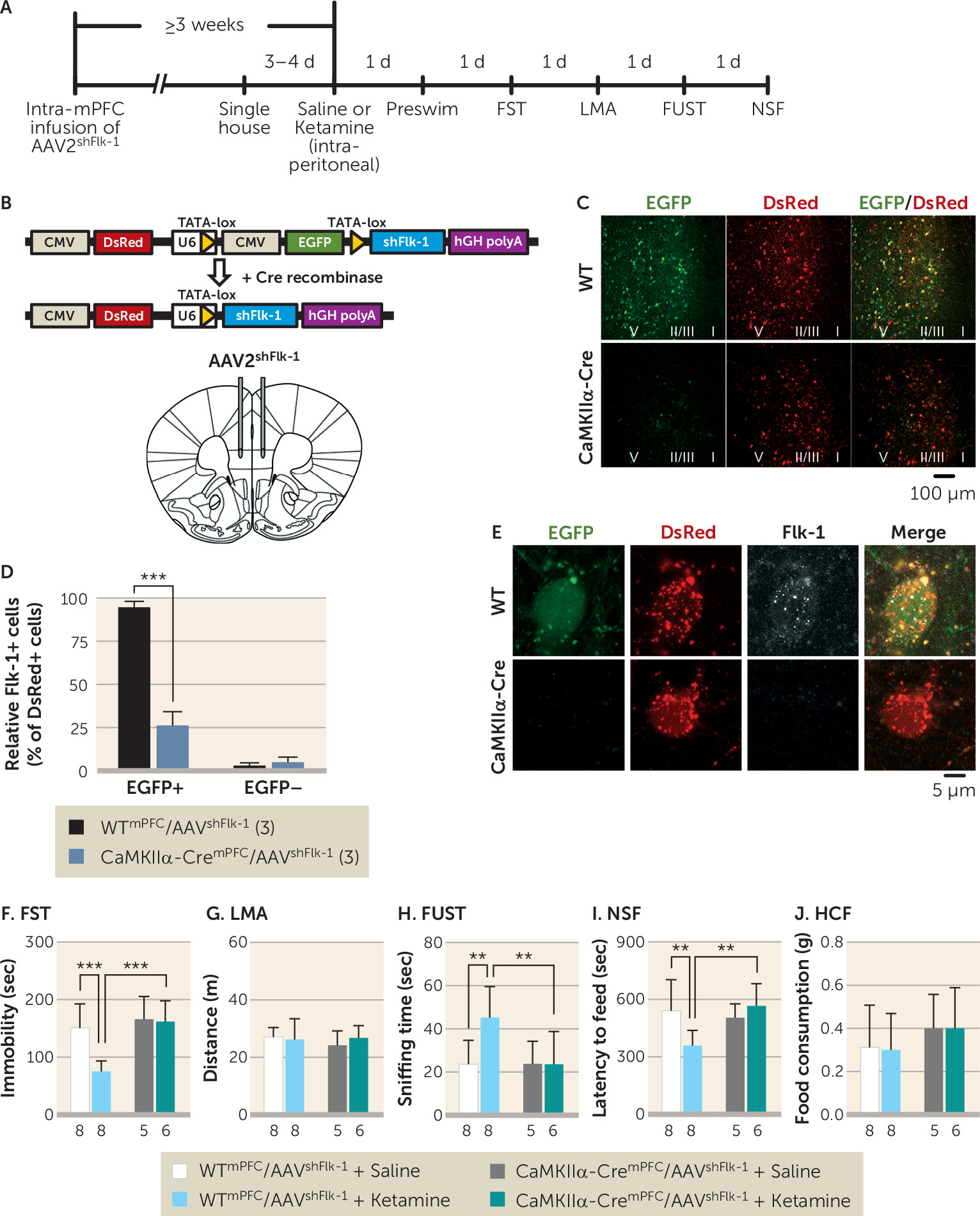
While VEGF–Flk-1 signaling is required for the actions of ketamine, the results demonstrate that blockade of neuronal VEGF–Flk-1 signaling did not result in increased immobility in the forced swim and tail suspension tests, increased latency to feed in the novelty-suppressed feeding test, or decreased time in the female urine sniffing test. These findings indicate that under controlled, environmental, nonstress conditions, loss of neuronal VEGF–Flk-1 signaling is not sufficient to produce these behavioral changes. Similar findings have been observed in BDNF mutant mice; only when these mice are exposed to stress is depressive vulnerability observed (
35–
37). This could be due to the simultaneous actions of multiple neurotrophic factors, such that loss of one (e.g., BDNF or VEGF) is insufficient to produce behavior deficits. However, a previous study reported that viral-mediated VEGF knockdown in the hippocampus increased immobility and latency to feed in the forced swim test and novelty-suppressed feeding test, respectively (
38); this could be due to nonselective VEGF knockdown, in glia and endothelial cells as well as neurons, whereas deletion in the present study was specific to neurons. The earlier study also reported that nonselective VEGF knockdown in the hippocampus reduced but did not block the behavioral actions of ketamine. Further studies are needed to determine whether blockade of both VEGF and BDNF signaling in neurons and/or glial and endothelial cells in the mPFC is sufficient to produce behavior deficits. In addition, studies are needed to determine whether knockdown of neuronal VEGF–Flk-1 signaling increases susceptibility to stress in rodent models, such as chronic unpredictable stress or social defeat (
21), and whether the behavioral actions of ketamine in these models require VEGF–Flk-1 signaling.
Neuron-specific deletion of Flk-1 or VEGF did not result in overt developmental phenotypes, although there were some subtle effects. Basal spine density in layer V pyramidal neurons was decreased, and open arm time in the elevated plus maze was reduced, in the VEGF
neuron−/− mice; in Flk-1
neuron−/− mice, preswim immobility was increased, latency to feed in the novelty-suppressed feeding test was decreased (in saline- and/or ketamine-injected mice), and center time in the open field test decreased. The mechanisms underlying these effects are unknown but may be related to neuronal Flk-1 deletion throughout the forebrain, as region-specific, viral-mediated Flk-1 knockdown in the mPFC had no effect on forced swim test or novelty-suppressed feeding behaviors. Moreover, these effects were not observed in VEGF
neuron−/− mice. Further studies using viral-mediated Flk-1 knockdown are needed to determine whether other forebrain regions could explain the complex phenotypes shown in the Flk-1
neuron−/− mice. Alternatively, these behavioral differences could be related to the housing conditions: single-housed Flk-1
flox/flox/Flk-1
neuron−/− mice were more active than VEGF
flox/flox/VEGF
neuron−/− mice in the locomotor activity and tail suspension test, but there was no difference in locomotor activity when these lines were group housed. Also, although Flk-1
flox/flox and VEGF
flox/flox mice were backcrossed onto C57BL/6J mice, there could still be genetic variation that could account for the locomotor differences. Notably, the Flk-1
flox/flox line includes background of the outbred ICR strain (
26) that shows higher locomotor activity in a novel environment than C57BL/6J mice (
39). Despite these baseline differences, the controls for both lines show responses to ketamine that are blocked in the littermate knockout mice, demonstrating the utility of these lines for our studies of neuronal VEGF and Flk-1. Taken together, the conditional, cell-specific knockout approach provides strong evidence that forebrain excitatory neurons are both the source and the target of VEGF that is released in response to ketamine.
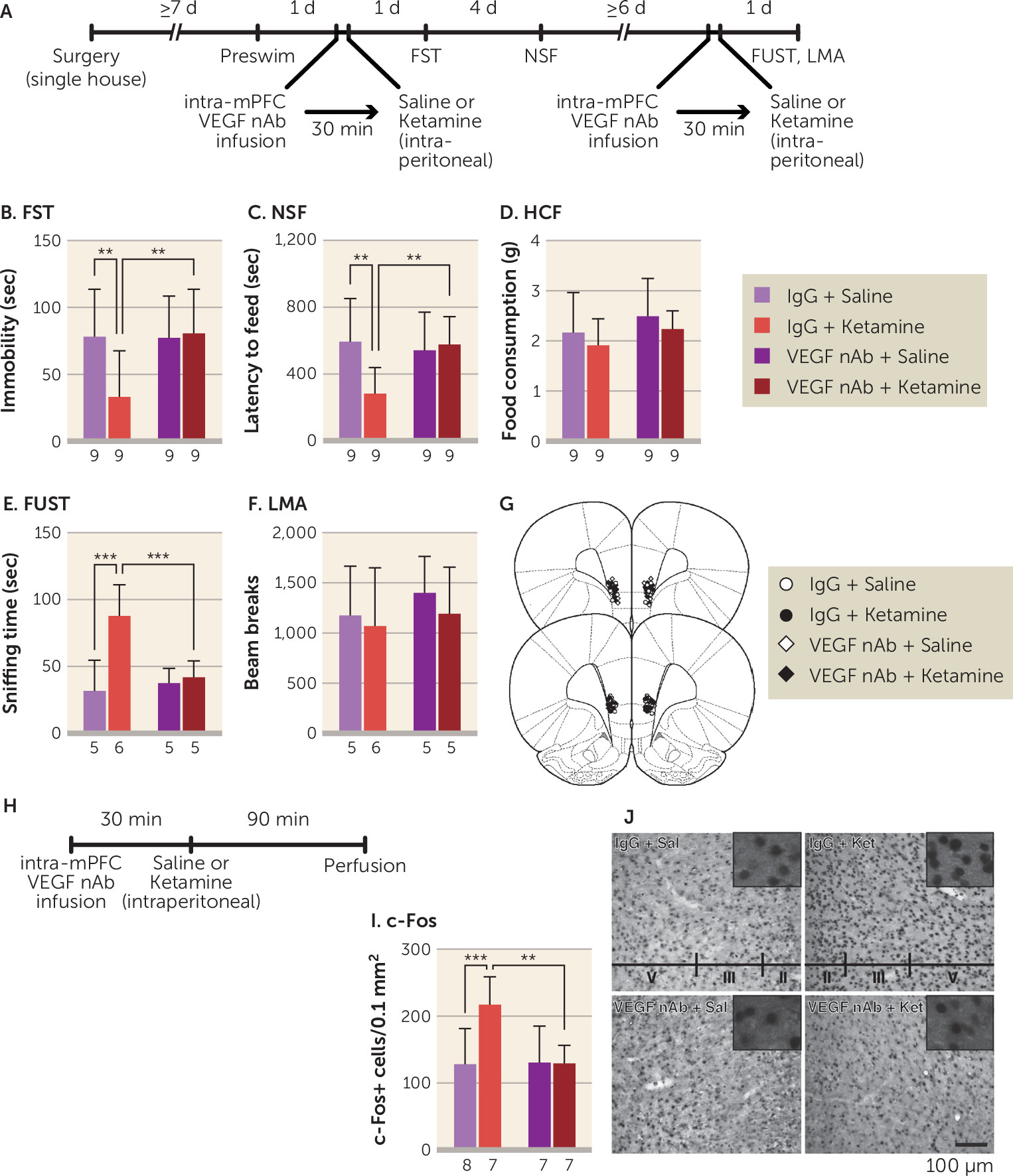
Analysis of c-Fos shows that VEGF-dependent neuronal activation in the mPFC is associated with the behavioral actions of ketamine. This is consistent with our previous results demonstrating that neuronal silencing of the infralimbic mPFC blocks the induction of c-Fos and the behavioral effects of ketamine and that in vivo optogenetic stimulation of the mPFC produces ketamine-like behavioral responses in the forced swim test, novelty-suppressed feeding test, and sucrose preference test (a test for reward or anhedonia) in rats (
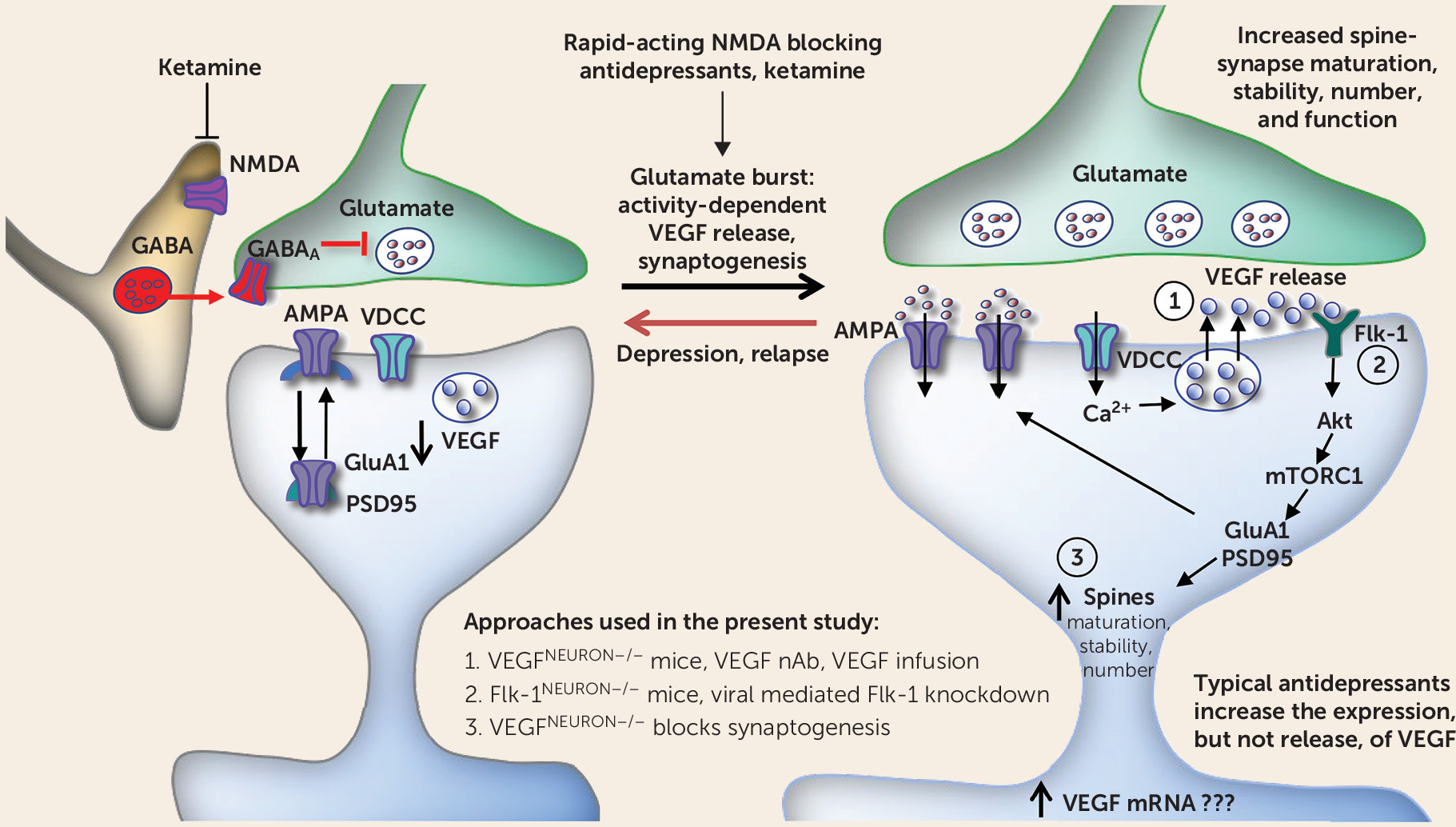
30). Consistent with these data, the present study shows that infusion of a VEGF neutralizing antibody into the mPFC prior to ketamine administration completely suppresses both the behavioral responses and c-Fos. In contrast, infusion of the VEGF neutralizing antibody into the mPFC 2 hours after ketamine administration did not block the behavioral effects of ketamine. Moreover, ketamine did not alter VEGF levels in the PFC within 1 hour. Taken together, the results indicate that ketamine rapidly and transiently increases VEGF release, but not protein levels, in the mPFC and that VEGF–Flk-1 signaling during the initial 2-hour period is required for the rapid and sustained behavioral actions of ketamine (
Figure 6). Previous studies report that ketamine increases levels of VEGF as well as BDNF in the hippocampus, but these studies did not test whether hippocampal VEGF or BDNF release is required for the actions of ketamine (
38,
40), as was done in the present study and in our previous work investigating BDNF (
22,
23). These findings indicate that rapid increases in VEGF and BNDF release, but not expression, in the mPFC at early time points less than 2 hours postinjection are critical for the behavioral effects of ketamine.
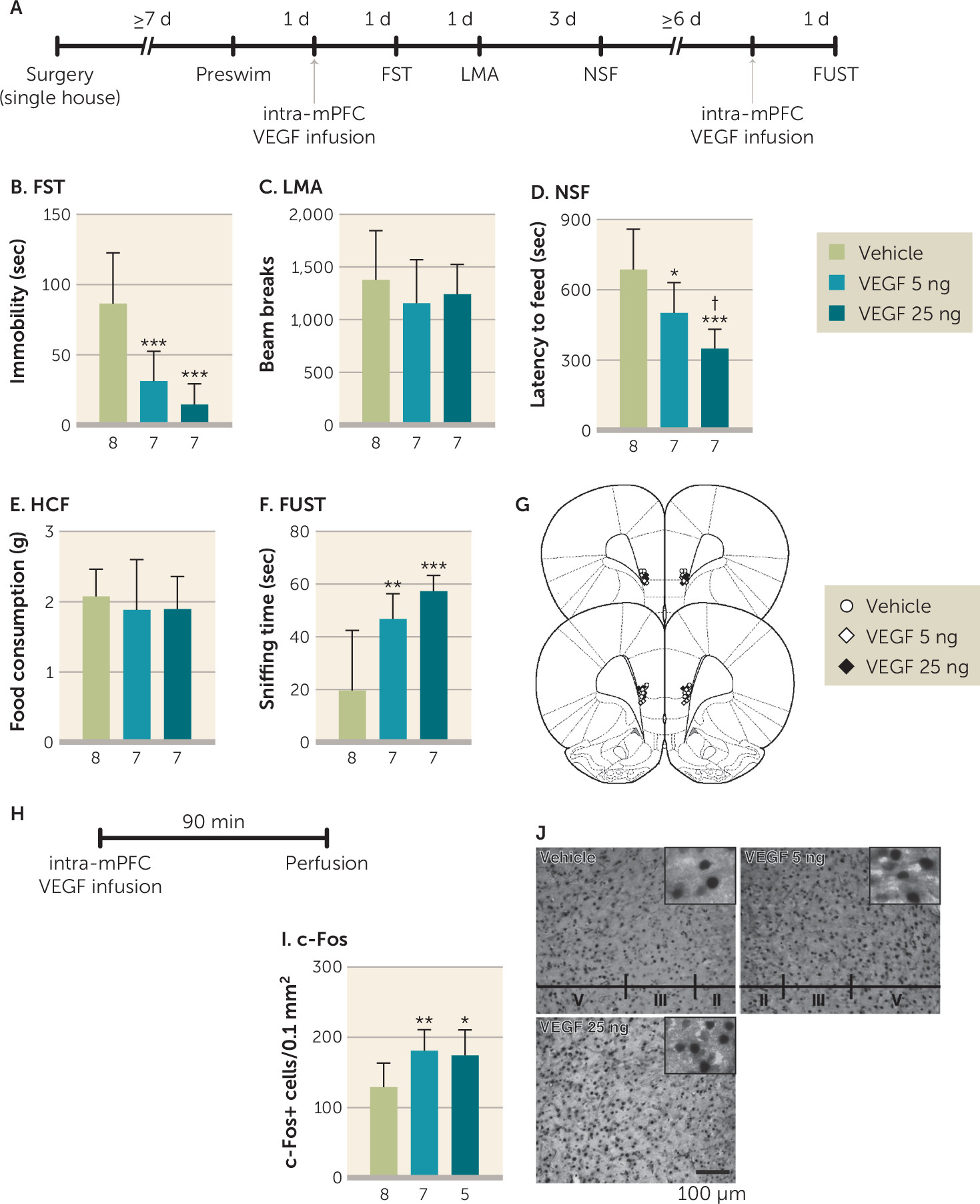
Previous studies have demonstrated that ketamine rapidly increases the number and function of spine synapses on layer V pyramidal neurons in the mPFC through activity-dependent BDNF release and activation of the mechanistic target of rapamycin complex 1 (mTORC1) (
5,
20,
23). The present results demonstrate that VEGF–Flk-1 signaling is sufficient to increase dendrite complexity in primary neurons, consistent with a previous report (
24). VEGF–Flk-1 is also necessary for ketamine induction of dendrite complexity and spine synapse number in layer V mPFC pyramidal neurons, as these effects were blocked by pharmacological inhibition of Flk-1 and in VEGF
neuron−/− mice, respectively. These results indicate that neuronal VEGF, as well as BDNF (
22,
23) signaling, play key roles in the synaptogenic effects of ketamine. In addition, spine density is decreased in the mPFC of VEGF
neuron−/− mice, similar to what is seen with chronic stress (
21,
41,
42), although these mice do not display behavior deficits in models of despair, anxiety, and motivation or reward. Similar morphological, but not behavioral, deficits (i.e., despair in the forced swim test) have been reported in BDNF Val66Met knock-in mice in which the secretion of BDNF is impaired (
23). These findings suggest that reductions of both VEGF and BDNF signaling are required to produce behavior deficits, as deletion or inhibition of only one pathway is insufficient. Whether these trophic factors act in parallel or sequentially remains to be determined. BDNF is reported to stimulate VEGF expression via mTORC1 in neuroblastoma cells, supporting the possibility that VEGF–Flk-1 signaling is downstream of BDNF (
43). However, it is also possible that ketamine induction of neuronal activity causes release of both BDNF and VEGF that then act simultaneously.
In summary, the present findings demonstrate that VEGF signaling in the mPFC plays an essential role in the behavioral and synaptic effects of ketamine and may have important implications for understanding the pathophysiology and treatment of depression. Although the role of VEGF in depression remains unknown, a recent genome-wide association study revealed that a VEGF-related single-nucleotide polymorphism (SNP), rs4416670, is associated with increased risk for depression (
44), and the minor C allele of this SNP is associated with decreased levels of VEGF (
45). Together, these findings raise the possibility that patients with the C allele and reduced levels of VEGF would not respond to ketamine to the same extent as patients with the major T allele SNP. Further studies of the impact of this SNP in humans as well as in rodent models could test this interesting hypothesis. In addition, the present findings could help identify novel VEGF–Flk-1 signaling sites to target for the development of new, rapid-acting antidepressants.