Why are genes important in psychiatry? The simple answer is the same reason they are important in other areas of biology and medicine—because most human traits and clinical disorders have a substantial genetic component. Genes influence and encode mechanisms of disease; they provide objective insights into individual predisposition or risk; they may help contextualize the interacting environment; and they are a potential tool for the discovery of treatments based on causation and pathogenesis.
It has been clear for more than a century that psychiatric syndromes run in families. Archival studies of concordance differences between identical and fraternal twins and occurrence of schizophrenia in individuals who were adopted away at birth and in their biological parents led to the conclusion that the main reason psychiatric diagnoses run in families is because of inheritance rather than culture or family dynamics. Psychiatry even before Freud has been essentially a phenomenological discipline, based on differences that can be observed between individuals with a particular diagnosis and those without one. Such phenomenology has been rich and compelling, with persuasive inferences about mechanisms from various schools of descriptive psychology and psychopathology and, more recently, evidence for biological associations from molecular studies of patient tissues and from neuroimaging studies. Causation, however, is not discernible from these phenomenological approaches. In contrast, genes are not merely phenomenological observations. They are the proverbial “egg” rather than the “chicken.” The study of gene function and genetic variation generates insights into etiological mechanisms and into the root causes of psychiatric syndromes.
Genes are also important because they make it possible to begin to unravel the critical contributory role of the interacting environment in complex disease. Genes are finite and quantifiable biological entities; the environment, by contrast, is potentially far more complex, and without an understanding of an individual’s discrete biological background (i.e., “genome”), the environment is a much more nebulous and indefinable entity. If life is imagined as a game of Monopoly, genes will never determine how many railroads one owns or where hotels will be built. But genes are the only square on the board that can be known with certainty—they are the “Go” square. Genes represent the biological tool box with which one plays the game, the underlying basis of individual characteristics that interact with the environment and lead to personal gain or resilience to loss. Daunting stress to one individual may be an exciting opportunity to another. As Paul Simon sang it, “one man’s ceiling is another man’s floor.”
There is abundant excitement in the research and pharmaceutical industry communities about the prospect of genetic discoveries leading to novel treatments based on elucidation of fundamental mechanisms of illness. Finding a disease-associated gene and translating that discovery into a therapeutic agent is a daunting challenge, and so far, in all of medicine, very few discoveries of disease-associated genes have led to novel treatments. In psychiatry, the treatments currently used, be it talk, electricity, magnetism, or pharmacology, were discovered through understanding of cause and pathogenesis; rather, nearly all were identified serendipitously. Having insights into mechanisms of psychiatric illness based on genetic information will surely be a more successful approach to therapeutic discoveries.
Discovery Of Schizophrenia Risk–Associated Genetic Variants
The landscape of genetic research in psychiatry has changed dramatically over the past decade. From family studies that were state of the art during the closing decades of the 20th century, the current fashion in medical genetics is the genome-wide association study (GWAS) comparing the DNA “genotypes” of clinical case subjects with control subjects. This approach is based on technological advances that allow inexpensive and high-throughput analysis of sequence variation across the human genome in DNA extracted from blood or saliva. Genotyping millions of common sequence variants, called single-nucleotide polymorphisms (SNPs), across the 3 billion nucleotides of a person’s genome can now be accomplished for little more than $100.
To facilitate discovery of genetic factors with rigorous statistical objectivity, based on no a priori expectation that any particular sequence variant will be associated with a particular trait or clinical illness, large samples have been acquired through collaborative consortia of investigators around the world. Indeed, GWAS has been applied to hundreds of traits and complex diseases to date, successfully yielding statistically significant risk-associated genomic regions for cancers, endocrinological disorders, and cardiovascular diseases, as well as for psychiatric illnesses, including schizophrenia, autism, mood disorders, posttraumatic stress disorder, and eating disorders.
The current state of the art in schizophrenia genetics is the product of the schizophrenia working group of the Psychiatric Genomics Consortium (PGC). In what is called the PGC2 schizophrenia study (
1,
2), which analyzed over 40,000 individuals with schizophrenia diagnoses and over 64,000 individuals identified as “controls” from over 50 centers in Europe and the United States, SNP allele frequencies that significantly differ between the patient and control samples have been identified in 145 regions (called “loci”) across the genome. Because millions of SNPs were surveyed for allele frequency differences between the samples, a statistical threshold for potential false positives was set at a conservative p value of 5×10
−8. These regions are presumed to contain genes that increase susceptibility to schizophrenia.
The finding of genome-wide statistically significant loci for schizophrenia has transformed the landscape of research in psychiatry, representing compelling evidence of the potential biological underpinnings of schizophrenia at a basic molecular level. The PGC2 study has recently been expanded (“PGC3”), and, as reported at the 2018 World Congress of Psychiatric Genetics in Glasgow, with 30,000 additional subjects, the number of GWAS-significant loci is now 246. As expected, most of the new loci were near statistical significance in the PGC2 version, confirming that increasing sample size will tend to elevate near statistically significant loci into the rarefied range of GWAS significance.
Some Caveats About Common Variants
While increasing the size of study samples has successfully provided increased power to identify loci that achieve statistical significance, larger samples do not increase the effect size, as reflected by the odds ratio of an individual risk-associated SNP. Even the SNPs with the greatest statistical significance increase risk on an individual basis only to a minuscule degree. Most of the SNPs increase risk less than 1.1-fold from general population risk (
1,
2). Moreover, the differences between patient and control samples in the frequency of alleles associated with risk is typically no more than 2%. Thus, by themselves, individual GWAS-significant SNPs have little or no role as predictors of a person’s clinical risk. As sample sizes grow even further, the odds ratios of newly significant GWAS loci will predictably be even smaller, and it is conceivable that within a few years, we will have in our gene quivers many hundreds of associations that increase risk fractions of a fraction. This is an important caveat to overly enthusiastic interpretations of existing data and promises of future GWAS analyses. It also is important to realize that the genetic analyses of this large case-control cohort characterize primarily individuals of European ancestry. Comparisons with other populations will require similar large studies of those populations, and preliminary evidence from Asian samples, although consistent to a moderate degree in terms of loci and effect sizes, has uncovered substantial divergences (
3). In African American samples, the attribution of population risk based on common alleles identified in European ancestry samples is likely to be much less consistent.
It is also important to appreciate that the risk loci identified are not themselves susceptibility genes. They are relatively low-resolution map points of regions of the genome that enable more detailed analysis to identify the specific genetic components accounting for the statistical association between DNA variation and clinical status. Most of the schizophrenia-associated loci contain more than one gene, and often contain many genes. From the physical gene map and the clinical statistics alone, it is impossible to conclusively identify the causative gene in any locus. This task is further complicated by the fact that most of the risk-associated SNPs are not in the coding portion of any known genes, meaning that they do not directly change the amino acids in the protein. Rather, they may be located within regulatory domains in the DNA that influence gene expression. While the exons of most genes encode proteins, the vast majority of the genome is noncoding. Recent work by the National Institute of Health ENCODE and National Institute of Mental Health PsychENCODE projects (
4,
5) has demonstrated that much of the noncoding genome is involved in regulating how genes are expressed, and individual noncoding variants in “enhancers” can influence the processing of genes many thousands of nucleotides from their physical location in the genome. In other words, because a SNP of interest lies near or even inside the physical boundaries of a gene does not necessarily mean that it is a factor in the biology of that gene.
Early Lessons From Common Risk Variant Discovery
Although the current data only scratch the surface of the underlying biology of risk, the schizophrenia GWAS results have arguably taught us a number of seminal lessons already. First, there is no single schizophrenia gene, per se. Individual SNPs indicate minor components of risk, not fate. Second, there is no royal road to risk. The many loci that are highlighted span the entire genome, involving every chromosome, and a schizophrenia-associated locus is likely to be found in virtually any 5 million base pair section of the genome. Indeed, there are many roads that potentially converge to contribute to risk, a characteristic of most so-called polygenic disorders. Third, the GWAS loci associated with schizophrenia are not unique to schizophrenia. Many of the same loci have been associated with other psychiatric diagnoses, including bipolar disorder, autism, and attention deficit hyperactivity disorder (ADHD) (
6,
7). It should come as no surprise that the human genome did not evolve to validate the DSM-5 or ICD-10 schema of psychiatric nosology. Fourth, many SNPs associated with schizophrenia appear to influence the epigenetic state of the genome, that is, the molecular machinery that regulates gene expression by changing chromatin conformation and methylation of DNA (
8–
10). Epigenetic factors respond to environmental events and underlie gene-by-environment interactions that are critical for development and aging, for learning, and for shaping behavior patterns based on experience. These studies suggest that many genetic variations related to risk for schizophrenia (and likely other psychiatric diagnoses) influence the nature and sensitivity of the genomic response to environmental events.
Fifth, most genes located within GWAS-significant loci are not genes that confirm earlier hypotheses about the biology of schizophrenia. Notwithstanding the aforementioned uncertainty of which, if any, genes within a locus are actually causative, there are a handful of genes in glutamate and GABA signaling pathways in GWAS loci, and one GWAS-significant locus contains the dopamine type 2 receptor gene (DRD2). Most of the genes within these loci, however, encode proteins involved in biological and molecular processes not previously emphasized as potentially related to schizophrenia or any psychiatric disorder. Many genes encode proteins in intermediate metabolism and the core processes of cell signaling and cell cycle biology, while many others reflect cellular functions about which very little is known. In an effort to reduce the potential biological complexity, if not the opacity, of the diverse functions involved and find common biological pathways of the sets of genes contained in the GWAS loci, investigators have applied bioinformatic tools, hoping to find some functional convergence. The assumption here is that despite the apparent biological diversity of the implicated genes, there may be common functions that underlie core phenotypes. The bioinformatic tools employed in these “in silico” studies give only crude approximations of biological reality, as they are based on the available research literature and databases and often on peripheral, but not CNS, biological data. Nevertheless, the results have suggested that some of the gene sets within schizophrenia GWAS-significant loci converge on several quite general aspects of cell biology, including calcium channel signaling, synaptic function, and basic neurodevelopmental processes (
11–
13). These results are consistent with the general idea that genes associated with a behavioral disorder influence the development of the brain and the plasticity of cells, particularly how they “talk to each other” and respond to environmental stimuli. However, as appealing as these results are, the validity and utility of conclusions about discrete pathogenesis or discrete pathophysiological mechanisms derived from bioinformatic gene set and pathway analyses should be interpreted with caution. The biological roles that genes play in brain function are highly context dependent, spatially (cell and tissue dependent), temporally (in terms of development and time of life), and in terms of background (what other genes are doing in the same cell).
Another lesson learned from the large-scale schizophrenia GWAS is that the heritability of schizophrenia—that is, the fraction of risk accounted for by common genetic variation as analyzed under simple additive models—is not explained by the cumulative associations reported to date, nor even by the GWAS results expected over the next decade. Indeed, the significant SNPs in the PGC2 study explain only a few percent of heritability, and predictions are that less than 30% will be explained by GWAS findings in the coming years (
1,
2). The reasons for this apparent “missing heritability” are unclear and the subject of considerable debate. One view holds that the model for calculating heritability is based on a simple linear addition of risk factors and does not accurately represent how genes influence biology. In most nonhuman contexts, including in yeast, worms, flies, and rodents (
14–
17), and also in human cancer cells (
18), genes interact in nonadditive ways—biological processes in living organisms tend not to be linear. Whether this is a major factor in the architecture of schizophrenia risk is unknown.
A recent and provocative hypothesis, the “omnigenic hypothesis” (
19), opines that heritability related to common genetic variation will eventually be more fully explained when sample sizes are in the millions, based on the assumption that variation in any gene expressed in a relevant cell type will contribute to risk. The omnigenic hypothesis argues in effect that any gene expressed in a neuron matters to the biology of that neuron, and because cell function depends on many layers of interacting protein networks, any gene expressed in a relevant cell will exert at least a small influence on the function of multiple networks in that cell. While some networks are presumed to contain more pathogenically critical, or “core” genes, most of the genes in GWAS loci will contribute to risk simply because they are expressed in the relevant cell, even though they are relatively peripheral to core pathogenesis. In other words, according to this hypothesis, variations in most genes expressed in neurons will contribute to the heritability of schizophrenia and eventually be in GWAS-significant loci (each with a trivial effect size) but will be of minimal biological relevance to understanding the pathogenesis of schizophrenia. This also is a controversial concept, in part because it is so challenging to the prevailing GWAS zeitgeist.
Rare Variants And Schizophrenia Risk
Genome-wide genotyping and, more recently, exome and whole genome DNA sequencing studies have discovered that approximately 2%−3% of patients with schizophrenia diagnoses have rare variations in their genomes that have deleterious effects on protein expression or sequence and in general have a greater influence on risk—that is, are more penetrant—than individual SNPs (
20–
22). These include relatively large DNA segments that are deleted or duplicated, called copy number variants (CNVs), and loss-of-function mutations of individual coding nucleotides. When DNA is copied, during meiosis or even during cell division, occasional errors occur, and sections of DNA may be duplicated or deleted, or single nucleotides may be mutated (single-nucleotide variants, or SNVs). If the mutation alters a protein or if the CNV is large enough, and typical risk-associated CNVs are on the order of 100,000 bases or more, genes within the altered region will be either deleted or duplicated. Because most of these more penetrant rare variants tend to arise during gametogenesis, they are typically found on one parental chromosome, so affected individuals are heterozygotic for the variation. It is thought that every person has at least one large CNV in their genome and probably several hundred protein-altering SNVs (
23). Most of these do not “cause” a clinical disorder. The most common CNV that consistently does so is the 22q11 hemideletion of approximately 3 million bases associated with the velocardiofacial syndrome, a condition in which approximately 30% of individuals will meet criteria for a diagnosis of schizophrenia in early adulthood. Large-scale studies of patients with schizophrenia diagnoses suggest that approximately 0.5% of individuals with this diagnosis have a 22q11 hemideletion (
21). A dozen or so other such recurrent CNVs have also been found to be more frequent in individuals with schizophrenia diagnoses than in control groups, but these CNVs have smaller effect sizes on risk (
21). Protein-altering SNVs that have been observed in patients diagnosed with schizophrenia are much rarer, and to date only one gene has been statistically confirmed as being a schizophrenia risk factor based on rare SNVs (
24).
It is important to note that the CNVs and rare SNVs associated with schizophrenia are not specific to schizophrenia but rather are also associated with other psychiatric and medical illnesses, including autism, ADHD, seizure disorders, and, in particular, intellectual disability. Indeed, individuals with schizophrenia-associated CNVs are more intellectually disabled than patients without these genetic variations (
25). Moreover, in the few reported cases of individuals with schizophrenia diagnoses who have mutations in
SETD1A, which is the only gene so far statistically confirmed as a risk factor based on a protein-altering SNV, a history of developmental disability (e.g., epilepsy and intellectual disability) is typical (
24). There is reason to question whether the psychosis in these cases is a primary phenomenon based directly on the genetic variant or a secondary associated feature often observed in individuals with intellectual deficits. It is unclear whether loss-of-function SNVs and large CNVs may be found in patients with schizophrenia diagnoses who do not have intellectual deficits, and claims to the contrary must be viewed with some skepticism (
25). Cognitive function is considered “normal” across a broad range, but for any given individual, being within that range does not mean that function is normal for that individual. As twin and family studies have shown, individuals with seemingly normal cognition and a schizophrenia diagnosis typically have poorer cognition than their siblings and other family members, even before clinical diagnosis (
26). The 22q11 hemideletion syndrome, the most penetrant CNV associated with “schizophrenia,” highlights the nosological ambiguity that underlies diagnosis of the schizophrenia syndrome. Recall that CNS syphilis was diagnosed as “schizophrenia” at the turn of the 20th century before the discovery of the spirochete. The velocardiofacial syndrome is associated not only with intellectual deficits but also with cardiac abnormalities, CNS malformations, and facial dysmorphia. Studies of psychosis in individuals with major intellectual disability, which used to be called “
pfropfschizophrenie,” have shown that the phenomenology is highly similar to that of patients with schizophrenia whose intellectual function is less impaired (
27). We are once again confronted with an epistemological conundrum because of the inaccuracies of our phenomenologically based syndromes. Furthermore, to the extent that many deleterious CNVs and mutations arise during gamete differentiation and are thus “de novo” and not inherited (i.e., not in the parental genome), it is unlikely that they contribute to missing heritability.
As if de novo mutations and segmental duplications and deletions in gamete formation did not add another non-Mendelian level of genetic complexity to human development, it has also become apparent that while every cell in a human body is a daughter of the fertilized egg, not every cell has the same genome. This is called somatic mosaicism. The same mistakes that can be made in DNA replication during meiosis can be made during cell division and perhaps even during gene expression when DNA is replicated or repaired. If these mosaic events occur early in development, they will characterize many subsequent daughter cells, but if they occur relatively late, for example, during the final stages of neuronal migration in the brain, they will affect only a small group of cells. The number of cells affected with a particular mutation or CNV is in essence a lineage marker for the history of that assembly of cells. Facial hemangiomas are a classic example of somatic mosaicism arising late in face maturation. Single-cell DNA sequencing studies in brain neurons have revealed that as many as 20%−30% of such cells have variations in their genome, including SNVs and CNVs (
28). While rare variations in individual neuronal genomes are found in the human brain, their role in CNS disorders, with rare exceptions (
29), has yet to be determined.
Beyond Schizophrenia Risk Variant Discovery
The landmark discovery of common and rare variants associated with schizophrenia has initiated a tidal wave of biological research to identify risk genes in the implicated genomic regions and elucidate specific disease mechanisms and disease pathways. Ongoing biological research, including analyses of gene expression, epigenetics, proteomics, and single cellular analyses, as well as animal modeling, has intensified as a result of these recent genetic discoveries. However, translating a GWAS positive locus into meaningful or actionable biology or a clinical translation or even a specific genetic entity is not straightforward and requires extensive further research. It is not a simple task to discover how a SNP identifying a GWAS locus relates to the biological activity of genes within the tissue thought to be of pathogenic relevance—the brain in the case of schizophrenia. Genetic variants that regulate expression can engage many molecular mechanisms accounting for their regulatory effects, such as transcription factor binding, small RNA interference, epigenetic variation, transcript splicing, and so on, but all these effects read out in the transcriptome. This is the rationale behind brain RNA sequencing studies being carried out in many centers around the world. As an illustration of the complexity of this problem, recent work has shown that even when association of the clinical risk SNP is made in brain to a particular expressed gene, the transcript showing association may not be the common, known form of the gene but rather an isoform not previously characterized (
30), or the classes of putative risk isoforms may vary across different brain regions (
31,
32), or the association in brain may depend on a particular time of life (
33) or perhaps even a specific cell type.
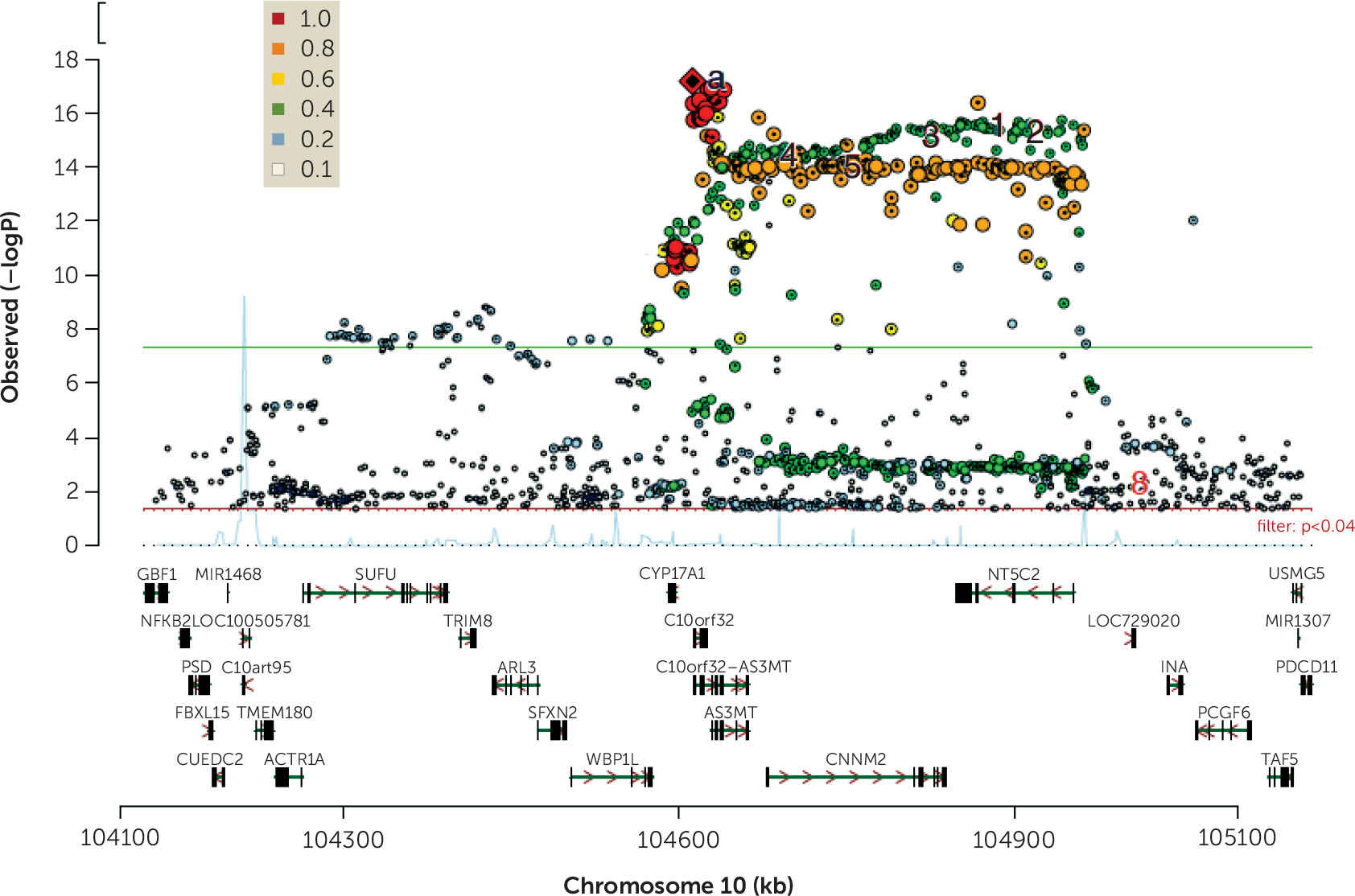
Figure 1 illustrates this problem for a highly statistically significant risk-associated locus, where many genes are mapped to the region. RNA sequencing and molecular characterization of transcripts in the region identified two genes underlying the clinical association—
BORCS7 and
AS3MT—and for
AS3MT, the transcript was a previously unknown, human-unique isoform of the gene (
30). To understand the molecular mechanisms of clinical association, precise characterization of the transcript association is necessary. For most GWAS loci, this degree of specification is not yet available.
In the meantime, while the biological research community engages the tools of functional genomics to elucidate the biological import of common and rare schizophrenia-associated risk variants, the clinical psychiatry community has begun to stratify patients by genetic risk and, in particular, by a cumulative sum of risk-associated variants across the genome, called the polygenic risk score. The clinical research landscape is beginning to be transformed by the availability of genetic risk factors in prediction models of risk, of environmental influence, and of outcome.
Polygenic Risk Score
One of the principal goals of genetic counseling is to advise about risk. As mentioned above for schizophrenia, rare genomic variants (CNVs) tend to be more penetrant compared with common genomic variants. The velocardiofacial syndrome deletion may increase risk of a diagnosis of schizophrenia up to 30-fold, and other identified rare variants associated with schizophrenia increase risk on average 5- to 10-fold. While this is potentially of value in clinical counseling, the odds are still much greater that schizophrenia will not occur in the context of these rare variants than that it will. Each common variant identified in GWAS, in contrast, increases risk only a tiny amount and thus cannot be used to predict risk. However, single variants are inherited on a background of millions of other variants in the genome, and genetic risk is presumably a cumulative manifestation of “genome type.”
In an effort to achieve a more meaningful prediction of genomic risk, a polygenic risk score (PRS) has been applied in many areas of medicine (
34,
35). The PRS is an approach to assigning to an individual a numerical score that quantitates his or her level of genomic risk for a particular trait. The PRS is calculated by summing all the alleles (weighted by their individual odds ratios) that have been associated with an illness in the latest GWAS data set for that illness. For example, for schizophrenia, considering SNPs in 145 independent loci contributing to risk in the current schizophrenia GWAS, a score can be assigned to an individual on the basis of how many of those specific risk-associated alleles for those SNPs are found in that individual’s genome. Scores can also be calculated from SNPs showing statistical association at varying p value thresholds (e.g., 5×10
−8, 1×10
−6, 0.05). In principle, two individuals could have the same score but actually share none of the same alleles that make up their score. The PRS is thus a gross aggregate measure of genomic risk, based on a simple additive model. Nevertheless, current PRS explains more risk than individual SNPs (on the order of 3%−15%, depending on which p value threshold is used [
34]). Moreover, because it is a score that can be calculated for any individual who has been genotyped, it has generated considerable interest as an independent measure for clinical research.
The PRS for schizophrenia has become a popular and dependable variable in differentiating groups of patients with schizophrenia diagnoses from control subjects, as patient samples have consistently higher scores than control samples. The schizophrenia PRS also tends to be increased in individuals with several other psychiatric disorders, particularly in the context of psychotic features (
6,
7). Use of the PRS has become a popular strategy in many studies as a predictor of associated clinical characteristics, such as cognitive deficits and treatment response, and it has also been associated with an expanding catalog of diverse measures and traits in unaffected people (e.g., adolescent cannabis use [
36] and body mass index [
37]). Perhaps not surprisingly, as a measure of overall genomic risk, the schizophrenia PRS is associated with aspects of early child development, including intellectual and emotional development and school performance (
6,
38–
40), as well as aspects of brain physiology associated with risk for schizophrenia (
41,
42). It is important to appreciate that associations and predictions in clinical samples based on PRS, although at times statistically strong, are still weakly predictive at the level of an individual and are not clinically actionable. Furthermore, PRSs to date are derived principally from samples of European ancestry, and questions have been raised about the potential confounding role of ancestral genetic components in mediating some of the clinical associations (
43).
The current incarnation of schizophrenia PRS has focused on index SNPs from the large GWAS study as an omnibus measure of genomic risk for clinical illness, but the concept can also be applied to basic biological data to identify genomic profiles that represent that biology in the clinical setting. For example, a PRS derived from a set of SNPs that influence expression of a network of genes co-expressed in brain with the dopamine D
2 receptor (DRD2) has been shown to predict response to antipsychotic drugs (
44). A PRS derived from a set of SNPs indexing genes in schizophrenia GWAS loci expressed in the human placenta and dynamically regulated in placenta from complicated pregnancies predicts the interaction of genetic risk and obstetrical complications on liability for schizophrenia (
45). The development of genomic profiles for specific biological processes has considerable potential for parsing the role of genes in biological processes that account for clinical variation. It is reasonable to hypothesize that schizophrenia PRSs deconvolved into “gene sets” reflecting specific molecular pathways or networks will have differential and stronger effects on specific clinical parameters than the whole-genome based PRS.
Comment
From a biological point of view, genetic research over the past decade has led to an unprecedented level of raw information about schizophrenia risk, as have genomic investigations of other traits and complex diseases across medicine, including in cancer, hypertension, diabetes mellitus, and cardiovascular disease. But how far will genetic insights take psychiatry? Will they change how patients are clinically diagnosed? Will they predict outcome and personalize medicine? Will they lead to novel therapies based on pathogenesis? My view is that the latter is a good bet, and the others are much more speculative. Given the substantial role of environmental variables in the etiology and course of all complex disorders, it is unlikely that even sequencing the DNA of everyone on the planet would lead to a full understanding of schizophrenia. It is important to continually emphasize that genes do not encode for psychopathology. Genes associated with schizophrenia implicate subtle malfunctions at the molecular and cellular level, most likely within neurons, altering micro- and macrocircuits in the developing brain. Cells process molecular information, and circuits process environmental information (e.g., sensory information, cognitive information, social information). Behavior is an emergent phenomenon of circuit physiology, which may be aberrant because the cells comprising the circuit are altered, for example, by not signaling to each other in an appropriate manner for a given context, based on the genetic and epigenetic programs that entrain their development and function. Psychiatric disorders ultimately reflect how the brain mishandles environmental information, which at the systems level is far “downstream” of the effect of genes in cells. Thus, given the complexity of the development and function of the brain and its emergent properties, determining what it means for a brain to be at increased risk for schizophrenia is a far more complex and challenging problem than finding susceptibility loci.
There is also reason to wonder to what degree GWAS associations sharpen the focus on the core pathogenic processes of schizophrenia. The omnigenic hypothesis mentioned above cautions that many GWAS associations are likely to be relatively peripheral factors in causation, detected because they play a role in the biological function of relevant cells. This may account in part for the substantial overlap between schizophrenia and not just other psychiatric diagnoses but even disorders as seemingly distant pathogenically as amyotrophic lateral sclerosis (
46). Moreover, while the loci that have been found seem not to respect our clinical diagnoses, the drugs we use to treat patients generally do. Lithium is not antipsychotic; clozapine does not benefit individuals with ADHD; and stimulants are not antimanic drugs. From a clinical perspective, treatment is a litmus test of biological relevance.
There is also concern that some of the genes within GWAS loci may not substantially contribute to symptomatology in adulthood. Studies of gene expression in adult postmortem brain samples of patients with schizophrenia do not generally find differential expression of genes within GWAS loci (
47,
48). In contrast, in a consistent emergent literature, many of the genes found in these loci have been found to be preferentially expressed and dynamically regulated in fetal life (
49), consistent with the prevailing assumption that schizophrenia, like most psychiatric syndromes, has early developmental origins (
50). A recent study (
45) found that a substantial fraction of genes in the GWAS-significant loci are abundantly expressed in the placenta and are dynamically regulated in placenta from complicated pregnancies, possibly explaining the link between schizophrenia risk and pregnancy complications. While this finding has potential implications for primary prevention based on enhancing placental health, it does not suggest specific treatment options for an affected adult.
These caveats notwithstanding, genes are critical and valid entry points to the biology of cells, and genes related to risk for schizophrenia are building blocks for model systems that may lead to novel insights and novel therapeutic targets. There is considerable enthusiasm in basic research laboratories for creating neuronal models based on human pluripotent stem cells from patients with schizophrenia and from cells with genomes that contain risk-associated variants (
51). In principle, such approaches may identify early developmental phenotypes that can be rescued experimentally. While this work also may not translate simply to the adult brain, it is worth remembering that the molecular programs that build synapses are at least in part the same ones that sustain them and modify them during adult life. Furthermore, animal research has shown that some early developmental abnormalities, even in brain structure, may be reversed during adulthood (
52). This is a new frontier, but it is not science fiction.
For good reason, 21st-century medicine has become highly intertwined with genomic-based medicine. Psychiatry is part of this new age, and the opportunities to change our nosology and our understanding of what we call mental illness and to find new approaches to improve the lives of affected individuals have never seemed more promising. That being said, as usual, the more we learn, the more we need to know, and with such a complex task, it’s likely to be a bumpy ride.
Acknowledgments
The author thanks Richard Straub, Rebecca Birnbaum, and Michael O’Donovan for their helpful suggestions and review of the manuscript.