Depressive disorders are common, chronic, and debilitating conditions characterized by affective, cognitive, and behavioral symptoms (
1). Anhedonia, a reduced ability to experience pleasure or a lack of reactivity to pleasurable stimuli, is a core symptom of depression and is associated with worse outcome, poor response to antidepressant medication, and increased risk for suicide (
2,
3). Oral medications for major depressive disorder approved by the U.S. Food and Drug Administration (FDA) largely consist of agents targeting the monoamine system. This lack of mechanistic diversity is likely an important contributor to the limitations in the efficacy of available treatments. Rational drug discovery based on a mechanistic understanding of disease pathology promises to deliver more effective, targeted therapies (
4,
5).
Dysfunction within the brain reward system is emerging as a core feature of depressive disorders, giving rise to deficits in response to pleasure, leading to anhedonia (
4,
5). The mesolimbic dopaminergic system plays an important role in the pathophysiology of depression (
6–
8). Preclinical studies utilizing chronic social defeat stress—a well-validated chronic stress model of depression—show that molecular and physiological perturbations within dopaminergic neurons projecting from the ventral tegmental area to the nucleus accumbens (NAc) (a component of the ventral striatum) characterize a susceptible, pro-depressive, and anhedonic phenotype (
8,
9). Within these brain regions, a critical role is played by KCNQ (or K
V7)-type voltage-gated potassium (K
+) channels, which includes KCNQ1-5. These channels are important regulators of cell membrane excitability and have been explored as potential targets of drug discovery for several CNS conditions (
10–
12). KCNQ2 and KCNQ3 form homodimers or heterodimers (e.g., KCNQ2/3 channels) and constitute the M-type channels (voltage-gated and ligand-regulated K
+ channels) that regulate neural excitability (
11). Critically, in the chronic social defeat stress model described above, mice that do not develop a depressive phenotype (i.e., resilient mice) show an up-regulation of voltage-gated K
+ channels, including the KCNQ3 channels within the ventral tegmental area, compared with mice manifesting the susceptible depressive-anhedonic phenotype (
8,
13,
14). Moreover, the susceptible phenotype can be reversed through overexpression of KCNQ3 channels, direct ventral tegmental area injection of various KCNQ channel openers, or peripheral daily administration of the selective KCNQ2/3 channel opener ezogabine, also known as retigabine (
13,
15).
Overall, these data provide a rationale for investigating the KCNQ2/3 channel as a target for drug discovery for disorders characterized by depression and anhedonia. As an initial proof-of-concept translational study, our group conducted a 10-week open-label pilot study of the effects of ezogabine, up to 900 mg/day, on clinical symptoms and brain connectivity in patients with major depressive disorder (
16). In that study, ezogabine showed good tolerability, and participants exhibited a significant improvement in clinical and behavioral measures of depression and anhedonia.
Here, we report the results of a two-site randomized controlled proof-of-concept exploratory trial testing the effects of ezogabine compared with placebo on brain response during reward anticipation and on clinical and behavioral measures of depression and anhedonia in adults meeting current criteria for a unipolar depressive disorder with elevated levels of anhedonia at baseline. This study was designed to test the KCNQ2/3 channel as a viable target for novel drug discovery for depression and anhedonia. Following an experimental therapeutics approach, the effect of treatment on brain response to reward was selected as a translational measure of target engagement. The primary outcome measure of the study was a change in activation during reward anticipation within the left and right ventral striatum from baseline to the primary outcome visit as measured by functional MRI (fMRI) during the incentive flanker task (
17). Secondary outcomes included clinical measures of depression and anhedonia.
Methods
Study Participants and Design
This was a multicenter, parallel, double-blind, randomized placebo-controlled clinical trial conducted at the Icahn School of Medicine at Mount Sinai in New York City and at Baylor College of Medicine in Houston. The institutional review boards at both institutions approved the study, and written informed consent was obtained from all participants prior to any study procedure. Study participants were recruited from web-based and newspaper advertising, as well as clinician referrals, between September 2017 and August 2019. Participants were compensated for their time and effort. Participants were between ages 18 and 65 and met DSM-5 (
18) criteria for major depressive disorder or persistent depressive disorder, as assessed by a trained rater using the Structured Clinical Interview for DSM-5–Research Version (
19). If a participant met concurrent criteria for both major depressive disorder and persistent depressive disorder in a current major depressive episode, major depressive disorder was indicated as the primary diagnosis. To be eligible to participate in the study, individuals could not be receiving any psychotropic medications at the time of randomization and had to exhibit clinically significant anhedonia with at least moderate illness severity, as defined by a score ≥20 on the Snaith-Hamilton Pleasure Scale (SHAPS) (
20) and a score ≥4 on the Clinical Global Impressions severity scale (CGI-S) (
21), respectively, at screening. Exclusionary diagnoses included any primary psychiatric diagnosis other than a depressive disorder as defined by DSM-5 (comorbid anxiety disorders and posttraumatic stress disorder were allowed), lifetime diagnosis of a major cognitive disorder, or substance use disorder in the past 6 months. Additional exclusion criteria are listed in the
online supplement.
After screening (28 days before and 1 day before baseline), individuals who met all eligibility criteria returned to the clinics to complete the baseline (day 0) assessments and undergo randomized assignment. Baseline assessments included clinical evaluations, the probabilistic reward task (
22,
23), and MRI scanning. Participants were then randomly assigned to one of the treatment arms (ezogabine, placebo) in a 1:1 ratio under double-blind conditions and entered the treatment period. Placebo tablets and ezogabine were encapsulated to preserve the study blind and were administered at a comparable frequency. During the treatment period, ezogabine was titrated according to FDA guidelines until reaching a maximum target dosage of 300 mg t.i.d. (900 mg/day) at week 4. This dosage was selected based on evidence showing that ezogabine reaches adequate brain levels and has demonstrated efficacy for seizure disorders at dosages between 600 mg/day and 1,200 mg/day (
24). According to the FDA package insert for ezogabine, the 900-mg/day dosage shows comparable efficacy and better tolerability compared with the 1,200-mg dosage for seizures. Moreover, in our pilot study, we utilized the 900-mg dosage, which demonstrated preliminary efficacy and good tolerability. The treatment period consisted of five study visits, which culminated in the primary outcome visit (study visit 5), where participants completed clinician-administered and self-report questionnaires and underwent the second and final MRI scan and probabilistic reward task assessment. At each visit, participants also completed self-report and clinician-administered rating scales performed by trained raters and met with a study psychiatrist, blinded to treatment assignment, who assessed suicidal thinking and behavior, adverse events, and changes in concomitant medications. Medication adherence was assessed throughout the study using pill count, participant diary, and participant report, as well as drug plasma levels on the primary outcome visit (study visit 5). Following this visit, participants were instructed to taper the study medication over 3 weeks in accordance with the FDA-recommended guidelines, during which they received weekly telephone calls from a member of the study team to assess adherence and side effects. After tapering, participants returned to the clinic for a final study exit visit. The total duration of participation was up to 14 weeks. (See Figure S1 in the
online supplement for a diagram of the study flow.)
The primary outcome measure of the study was a change in activation during reward anticipation (reward > neutral) within the left and right ventral striatum from baseline (study visit 0) to the primary outcome visit (study visit 5) as measured by fMRI during the incentive flanker task (
17). Secondary outcome measures were clinical symptoms of depression and anhedonia. Depression severity was measured using the clinician-rated Montgomery-Åsberg Depression Rating Scale (MADRS) (
25) and the Quick Inventory of Depressive Symptomatology–Self Report (QIDS-SR) (
26). Anhedonia severity was assessed using both the SHAPS, a validated 14-item self-report questionnaire that focuses on hedonic responses (
20), and the Temporal Experience of Pleasure Scale (TEPS) (
27), a validated self-report that yields specific subscores of anticipatory pleasure and consummatory pleasure. In addition, the degree of change in interest or pleasure from a specific experience relative to the past was explored with the Specific Loss of Interest and Pleasure Scale (SLIPS) (
28), while anhedonia related to social situations was explored with the Anticipatory and Consummatory Interpersonal Pleasure Scale (ACIPS) (
29). Global illness improvement and severity were evaluated at each visit by a study clinician using the CGI-S and the CGI improvement scale (CGI-I) (
21). Suicidal ideation and behavior were measured using the Columbia-Suicide Severity Rating Scale (C-SSRS) (
30). Adverse events were summarized in accordance with the Medical Dictionary for Regulatory Activities system organ class and preferred terms. Safety and tolerability were assessed by discontinuation rate, frequency of adverse events, and change in score on the C-SSRS. Clinical raters completed standardized rater training at each site and achieved interrater reliability >90% for the MADRS. Additional secondary outcomes included change in behavioral measures of reward learning through a computer-based task, the probabilistic reward task. Methods and results of ezogabine compared with placebo on the probabilistic reward task are presented in the
online supplement.
MRI Acquisition and Processing
MRI data were acquired in a Siemens 3-T MAGNETOM Skyra scanner with a 32-channel head coil at the Icahn School of Medicine at Mount Sinai, and in a Prisma scanner with a 64-channel head coil at Baylor College of Medicine. Scans included an anatomical T
1-weighted scan and a task-based functional scan with the incentive flanker task. The anatomical T
1-weighted images were acquired with a magnetization-prepared 2 rapid gradient-echo sequence (MP2RAGE), which collects two volumes after each inversion for improved image quality (TR=4,000 ms, TE=1.9, inversion 1/2 time=633/1,860, field of view=186×162, voxel resolution=1×1×1 mm), and the functional scan for task performance was acquired with a multiecho, multiband, accelerated echo planar imaging sequence (TR=882 ms, TE=11.0, 29.7, 48.4, 67.1, multiband factor=5, field of view=560×560, voxel resolution=3×3×3 mm, flip angle=45°). Functional scans were preprocessed and denoised for motion and physiological noise using multiecho independent component analysis. Multiecho fMRI data were decomposed into independent components and scaled against echo time. Components with high echo-time dependence are considered blood-oxygen-level-dependent (BOLD)-like, whereas components with low echo-time dependence are considered noise-like (
31,
32). Removal of non-BOLD components allows robust data denoising for motion, physiological, and scanner artifacts (
31,
32).
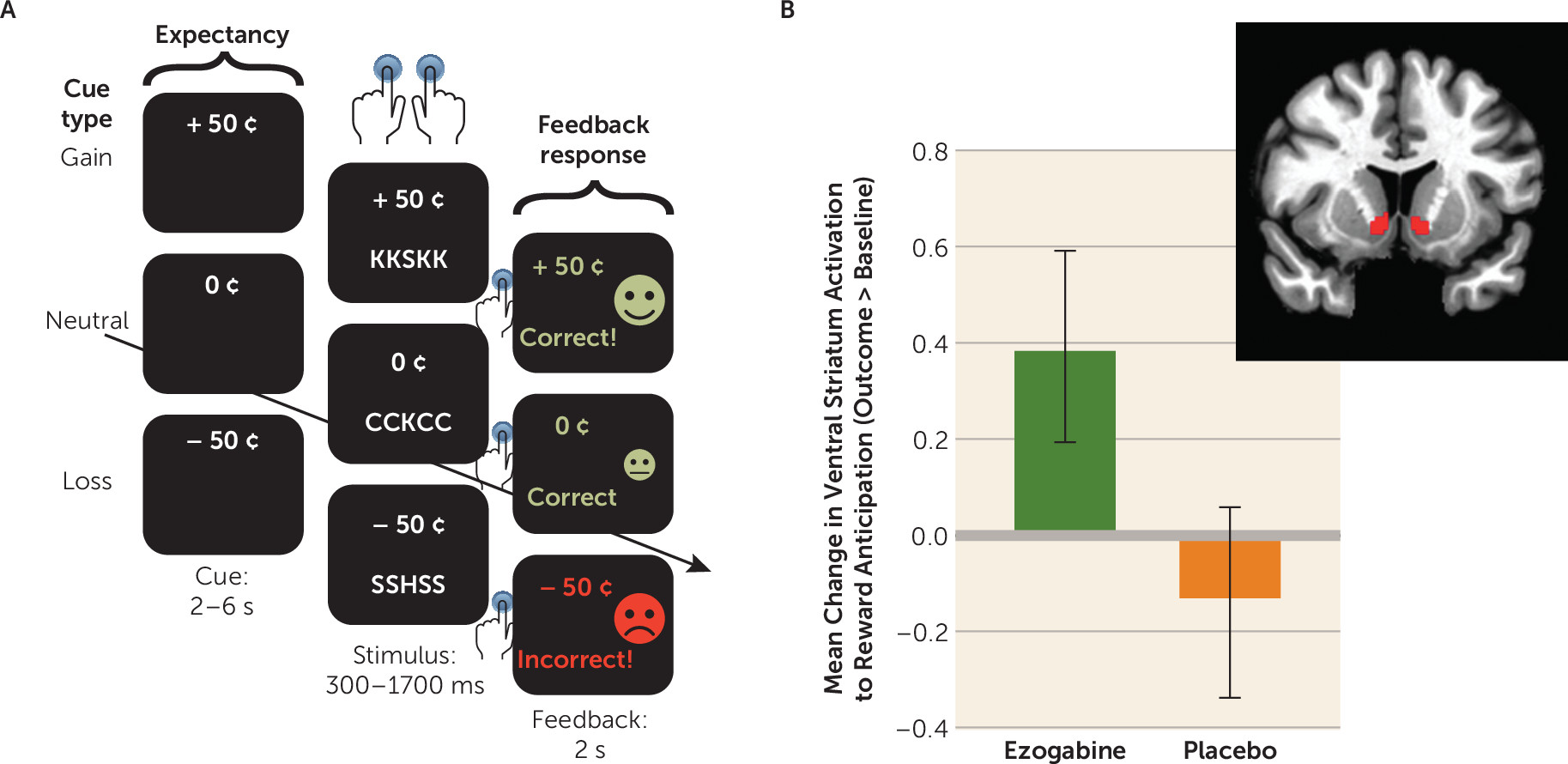
Incentive Flanker Task During fMRI
The incentive flanker task, like the monetary incentive delay task (
33), permits discrete modeling of brain activity during anticipation of an incentive (
17). Each trial begins with a monetary cue (reward, loss, or neutral), followed by a string of five letters. Participants were instructed to press one of two buttons to identify the center letter. Success requires accurate identification of the target within a specific response period, titrated based on performance during a practice session. Feedback on performance followed, including text reading “Correct!” or “Incorrect!” and the total value of any money won or lost. A total of 120 trials (40 each of reward, loss, and neutral) were presented in pseudorandom order, equally divided across four runs of approximately 6 minutes each. Task-based modeling was conducted using Analysis of Functional NeuroImages (AFNI) (
34) and the FMRIB Software Library (FSL) (
35) software. Subject-level general linear models included regressors for onsets for cue type (reward, loss, or neutral), flanker stimuli, and feedback. The duration of each cue varied between 2 and 6 seconds and was modeled with AFNI’s
dmBLOCK function and convolved with the hemodynamic response function. As noted above, the primary outcome for the study utilized reward anticipation computed as the contrast of the reward cue compared with the neutral cue (reward > neutral). The loss cue was contrasted against the neutral cue in order to explore whether the effects were specific to anticipation of potential reward. Parameter estimates for these contrasts were extracted within the ventral striatum region of interest (from the Harvard-Oxford subcortical structures atlas provided by FSL, labeled “nucleus accumbens”) (
35) for each subject and entered into statistical analyses. A description of the task is presented in the
online supplement.
Statistical Analysis
On the basis of pilot data, a total sample size of 48 was chosen to provide 85% power to detect a between-group difference in means of 0.8 standard deviations assuming a correlation of 0.6 between a pair of measurements made in the same participant. The primary imaging outcome and all clinical measures assessed at multiple time points were analyzed using linear mixed-effects models with a single random intercept term treating time as discrete or continuous, as appropriate. Adverse event rates were compared between treatment groups using Poisson regression. Continuous baseline clinical and demographic characteristics were summarized using means or medians and compared by group using t tests or Wilcoxon tests, as appropriate. Discrete baseline characteristics were summarized by count and percentage and compared using a chi-square test or Fisher’s exact test as appropriate. Correlation between measures was computed using Pearson’s r and Spearman’s rank correlation for parametric and nonparametric distribution, respectively. A two-sided significance level of 0.05 was set for all comparisons. No correction was made for multiple comparisons. Effect sizes for differences in mean change from baseline to outcome between treatment arms were computed as Cohen’s d (
36). Sensitivity analyses examined the effect of missing data by using the highest observed value and the lowest observed value for all missing primary outcome observations. All analyses were conducted on an intent-to-treat basis, using SAS, version 9.4 (SAS Institute, Cary, N.C.).
Results
Sample Characteristics
Seventy individuals were assessed for eligibility; of these, 45 were eligible for randomization and entered into the double-blind trial and represent the intent-to-treat analyzed sample. Enrollment ended with 45 participants who were randomly assigned prior to reaching the targeted sample size of 48 participants, which was a result of exhaustion of the available supply of the study drug. The clinical and demographic characteristics of the study sample are summarized in
Table 1. Dropout rates were low and did not differ significantly between groups; of the 45 randomly assigned participants, 42 completed the primary outcome, yielding a retention rate of 93.3%.
Across the whole sample, adherence to the study medication as measured by pill count was 96% at the primary outcome visit (study visit 5) and was >95% at each study visit. Blood samples were collected at the primary outcome visit to determine plasma levels of ezogabine and N-acetyl-ezogabine. For further details, see the online supplement, including Figure S2.
fMRI Data
Of the 45 participants enrolled, all had a pretreatment MRI scan, and 40 (88%) had both a pretreatment and a posttreatment fMRI scan. fMRI data during the incentive flanker task were processed and analyzed for the 40 participants for whom valid pretreatment and posttreatment data were available. Three participants did not complete the study primary outcome, and two participants were excluded because of failure to make any responses during the task. At baseline, the study groups did not differ in their performance accuracy (percentage errors in the ezogabine group, 10.4%, SD=10.3; in the placebo group, 9.5%, SD=8.3; p=0.77). Likewise, there was no difference in performance accuracy at the outcome visit (percentage errors in the ezogabine group, 8.1%, SD=8.2; in the placebo group, 6.1%, SD=5.1; p=0.51).
The effect of treatment on change in ventral striatum activation in response to reward from baseline to outcome was not significant. Participants in the ezogabine group showed a numerical increase in ventral striatum activation following treatment compared with those in the placebo group (estimate=0.52, SEM=0.28; t=1.85, df=38, p=0.07) (
Figure 1). The effect size (Cohen’s d) for the difference in change in means was 0.58. An exploratory analysis adding site as a random effect did not alter the results (estimate=0.53, SEM=0.28, df=38, p=0.07). There was no significant group difference in ventral striatum activation during loss of anticipation from baseline to outcome (p=0.73). Exploratory whole-brain-corrected analyses revealed no significant clusters for the interaction of drug by time (controlling for site, age, and sex; whole-brain cluster-corrected family-wise error corrected p<0.05, voxel-wise error level, p<0.001, K >25).
Symptom Change
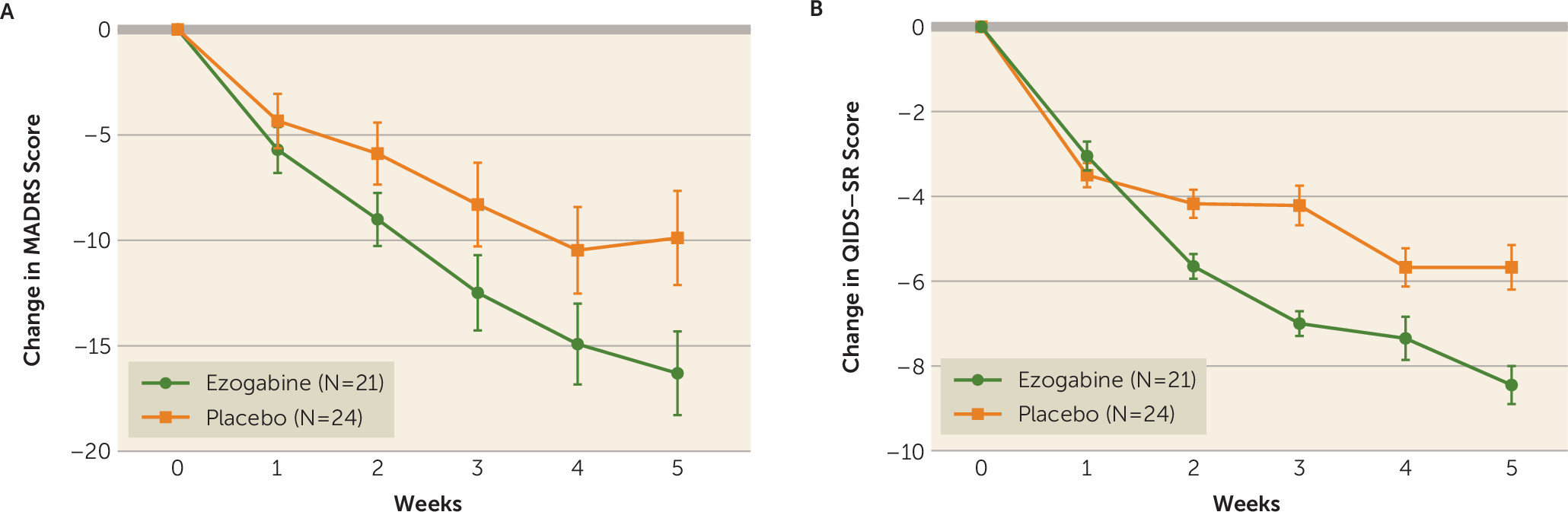
Ezogabine, compared with placebo, was associated with a large improvement in depression as measured by the MADRS (t=−4.04, df=213, p<0.001) (
Table 2,
Figure 2). The mean change in MADRS score from baseline to endpoint was 15.9 (SD=8.9) and 8.0 (SD=10.4) for the ezogabine and placebo groups, respectively. The mean difference in MADRS score between baseline and the primary outcome visit was −7.9 (SD=3.0), and the effect size for the difference in change in the mean for MADRS score was 0.76. The response rate (a reduction ≥50% in MADRS score from baseline to the primary outcome visit) was 61.9% (N=13/21) and 37.5% (N=9/24) for the ezogabine and placebo groups, respectively. The remission rate (MADRS score <10 at the primary outcome visit) was 38.1% (N=8/21) and 20.8% (N=5/24) for the ezogabine and placebo groups, respectively. The number needed to treat based on the remission rate was 6, and the number needed to treat based on the response rate was 4.
Similarly, compared with placebo, ezogabine was associated with a significant improvement in depression as measured by the QIDS-SR (t=−3.1, df=213, p=0.002). The mean difference in QIDS-SR scores between baseline and the primary outcome visit was −3.0 (SD=0.7), and the effect size for the difference in change in the mean for the QIDS-SR score was 0.56 (
Figure 2).
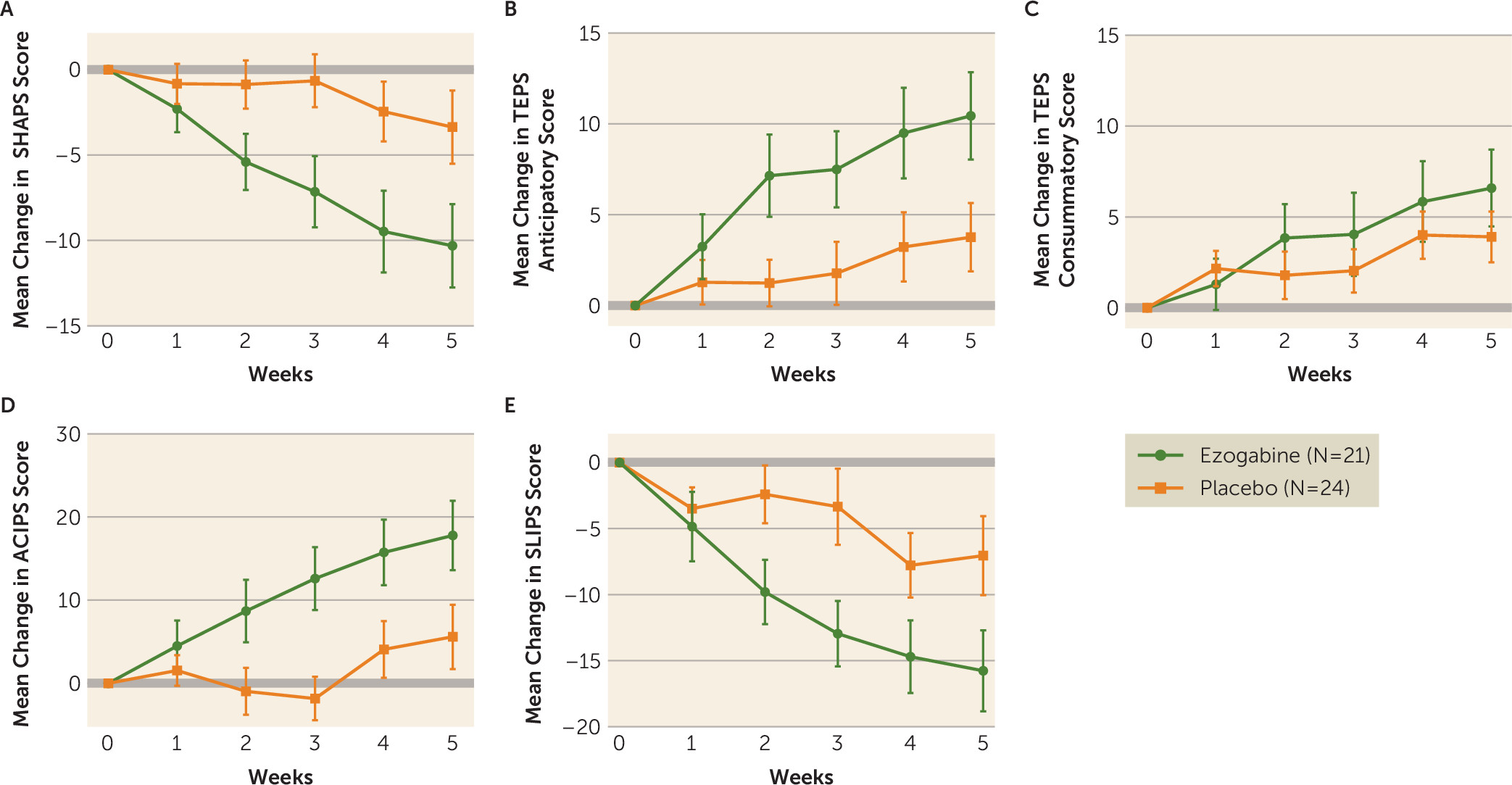
Compared with placebo, ezogabine was associated with a large improvement in hedonic capacity as measured by the SHAPS (t=−4.1, df=212, p<0.001) (
Table 2,
Figure 3) and in the ability to anticipate pleasure as measured by the anticipatory subscale of the TEPS (t=3.4, df=213, p<0.001) (Table,
Figure 3). A smaller benefit for ezogabine over placebo was observed for the consummatory subscale of the TEPS (t=2.0, df=213, p=0.05) (
Table 2,
Figure 3). The mean differences in scores on the SHAPS, on the anticipatory subscale of the TEPS, and on the consummatory subscale of the TEPS between baseline and the primary outcome visit were −6.9 (SD=3.2), 6.7 (SD=3.1), and 2.7 (SD=2.5), respectively. The associated Cohen’s d showed a large effect size for the difference in change in the means for the SHAPS and the anticipatory subscale of the TEPS (d=0.64 and d=0.66, respectively) and a small to medium effect on the consummatory subscale of the TEPS (d=0.34). Significant benefit for ezogabine over placebo was also observed for anhedonia as measured by the ACIPS (t=3.8, df=213, p<0.001) and the SLIPS (t=−3.4, df=213, p<0.001). Among participants in the ezogabine group, change in ventral striatum activation in response to reward anticipation from baseline to outcome was correlated with change in anticipatory anhedonia as measured by the TEPS (rho=0.49, p=0.04), such that increased ventral striatum activation was associated with increased self-reported anticipation of pleasure. This association was not observed across the whole sample (rho=0.12, p=0.45).
Finally, ezogabine, compared with placebo, was associated with a significant benefit in terms of both global illness severity (CGI-S, t=−2.2, df=214, p=0.026) and global illness improvement (CGI-I, t=−2.9, df=214, p=0.004). Details on clinical outcomes are presented in
Table 2.
Safety and Tolerability
No serious adverse events occurred in the course of the trial. The most common adverse events were dizziness and headache. Only two adverse events reached statistical significance (dizziness and confusion), and both were higher in the ezogabine group. In the ezogabine group, the most common adverse event was dizziness, which had an incidence rate of 39.7/100 patient-months. In the placebo group, the most common adverse events were headache (12/100 patient-months), dizziness (8.6/100 patient-months), and nausea (8.6/100 patient-months). Because of the occurrence of adverse events, four participants in the ezogabine group did not achieve the highest dosage (900 mg/day) and remained at 750 mg/day (N=2), 600 mg/day (N=1), or 450 mg/day (N=1); however, no participant in the ezogabine group withdrew from the study because of adverse events. One case of retinal abnormality (operculated retinal hole) not described at screening was reported at an ophthalmologist visit during the study exit for one participant in the ezogabine group. No elongation of the QT interval (operationalized as a QT interval over 500 ms or a 60-ms increase in the QT interval from baseline) was observed during the study. In the overall sample, there was no emergence of serious suicidal ideation compared with baseline, as defined by an increase in the maximum suicidal ideation score ≥3 on the C-SSRS during the trial. No participants experienced emergence of suicidal behavior during the study. A summary of adverse events in the study is provided in Table S1 in the online supplement.
Discussion
We report the results of the first randomized controlled trial, to our knowledge, investigating the effect of a KCNQ2/3 channel opener on brain and clinical measures linked to depression and anhedonia. The primary neuroimaging endpoint of the study, the effect of treatment with ezogabine compared with placebo on ventral striatum response to reward, was not met. Treatment with ezogabine compared with placebo was associated with significant improvements in clinician and self-report measures of both depression and anhedonia, as well as on measures of global illness severity. Improvement in response to reward anticipation within the ventral striatum was positively associated with improvement in self-reported anticipatory pleasure among participants treated with ezogabine, but not placebo. Ezogabine was generally well tolerated, and no serious adverse events occurred during the trial.
Following an experimental medicine approach, we selected the effect of treatment with a KCNQ2/3 channel opener on brain response to reward as a translational measure of target engagement. Preclinical studies highlight KCNQ channels as a potential novel drug target for the treatment of depressive disorders and anhedonia (
13,
14). Within the context of the chronic social defeat stress model described above, up-regulation of KCNQ3 channels is observed within ventral tegmental area dopamine neurons of mice resilient to stress, and genetic and pharmacologic enhancement of KCNQ3 channel function in these neurons both normalizes pathological hyperactivity of these dopamine neurons and reverses depressive behaviors in susceptible mice. Hyperactivity within the ventral tegmental area-NAc circuit in susceptible mice appears to be driven by intrinsic changes in channel function that lead to a hyperpolarization-activated cation channel-mediated current. In contrast, resilient mice are characterized by a stable firing that is homeostatically maintained by K
+ channels. As demonstrated by Friedman et al. (
13), KCNQ channels contribute to K
+ channel activity in resilient mice. The direct potentiation of KCNQ2/3 channels in susceptible mice was able to normalize the pathogenic hyperactivity of ventral tegmental area dopaminergic neurons and produce antidepressant effects at the behavioral level. A recent study by Feng et al. (
37) replicated these findings using a chronic, high-fat diet-induced model of neuroinflammation that results in depressive-like behavior.
In line with the Research Domain Criteria initiative put forward by the National Institute of Mental Health, we selected a specific neurobehavioral domain of functioning relevant to depression as the dependent outcomes for our study, namely, the positive valence system that encompasses reward anticipation. Depressed individuals show deficits in the capacity to anticipate pleasurable stimuli (
38), as assessed through self-report scales and laboratory-based measures (
39). Individuals with depression show abnormal responses to reward expectancy within a well-defined cortico-striatal reward circuit encompassing the ventral striatum (and the NAc), ventral tegmental area, and prefrontal cortex (
40–
42). These altered responses appeared to normalize with antidepressant treatment in several small studies (
43,
44), consistent with the preclinical work described above. A recent randomized controlled trial of the selective kappa opioid receptor antagonist aticaprant (JNJ-67953964) demonstrated a significant increase compared with placebo in ventral striatum activation during reward anticipation using the monetary incentive delay task and fMRI in patients with anhedonia across a spectrum of mood and anxiety disorders (
45). The present study utilized a similar methodology, including the use of fMRI with the incentive flanker task (a reward task similar to the monetary incentive delay task) to measure the hypothesized effect of treatment on the reward circuit operationalized with a region-of-interest-based approach focusing on the ventral striatum. In contrast to that study by Krystal et al. (
45), our study failed to show a statistically significant effect of treatment on the ventral striatum. In addition, while our study did show beneficial effects of treatment on several secondary clinical outcomes, the clinical benefit of the intervention in the Krystal et al. study was less clear. There are important differences between the two studies, which may have contributed to the somewhat divergent findings. Regarding the discrepancy of the effect of treatment on ventral striatum response to reward, it should be noted that our sample size was approximately one-half that of the Krystal et al. study, raising the possibility that the lack of a significant finding in our study represents a type II error. In fact, the magnitude of the effect of treatment on ventral striatum response was similar in both studies.
This study has several limitations. The primary limitation is the small sample size. As noted above, our negative finding on the primary outcome may reflect a type II error because the effect went in the hypothesized direction, it only narrowly missed the alpha threshold of 0.05, and the significant effects of treatment on the clinical outcomes suggest that the intervention may have activity at depression-relevant brain targets. Our study suggests that investigators should conduct studies powered to detect the effects of treatment on brain targets in a range that includes 0.58 standard deviation units because this was the effect size observed in both our study and the study by Krystal et al. Investigators utilizing limited sample sizes may consider alternative statistical approaches, such as utilizing a more lenient alpha level, a futility design, or a Bayesian approach. Other limitations of our study include the enrollment of medication-free participants experiencing a broader unipolar depressive phenotype with clinically significant anhedonia that can limit the generalizability of the findings. Additionally, the short treatment duration (4-week titration plus 1 week at target dosage) precludes conclusions regarding longer-term efficacy or tolerability. Finally, because the study lacked a healthy nondepressed control group at baseline, it is unknown whether patients in the trial had abnormally low ventral striatum activity in response to reward prior to treatment.
In conclusion, this is the first randomized placebo-controlled trial to investigate the antidepressant effect of the KCNQ2/3-selective channel opener ezogabine in patients with unipolar depression. Given the study findings, larger randomized controlled trials of KCNQ2/3 channel openers in mood disorders are warranted to explore their potential as viable treatments for depression and anhedonia.
Acknowledgments
The authors thank the research pharmacists at the Icahn School of Medicine at Mount Sinai and Michael E. Debakey VA Medical Center, including Ivy Cohen, Biju Johnson, Alla Khodzhayeva, and Giuseppe Difiore, for their extensive work on this project.