Gabapentin, a safe and well-tolerated medication that is approved by the U.S. Food and Drug Administration to treat postherpetic neuralgia, partial seizures, and restless legs syndrome, has garnered considerable attention for its efficacy in mitigating alcohol withdrawal symptoms (
1) and preventing relapse drinking in individuals with alcohol use disorder (AUD) (
1–
3). Although much research has focused on the neurobiological mechanisms of gabapentin’s (“on-label”) anticonvulsant, anxiolytic, and analgesic effects, few studies have focused on the mechanisms of its (“off-label”) therapeutic effects in AUD, which likely diverge as a result of the interacting effects of drinking and withdrawal on the brain (
4). The present study represents the first controlled investigation of the neurobiological mechanisms of gabapentin in people with AUD.
Although gabapentin was designed as a structural analogue to γ-aminobutyric acid (GABA), it is inert at GABA receptors and synapses (
5). Instead, gabapentin binds with high affinity to the α2δ-1 protein and exerts its primary therapeutic effects through selective blockade of presynaptic voltage-gated calcium channels containing the α2δ-1 subunit (
5). Recent research has revealed that gabapentin’s therapeutic effects may also involve 1) α2δ-1 interaction with non-calcium-channel proteins, including
N-methyl-
d-aspartate (NMDA) receptors, neurexin-1α, and thrombospondin (
6), as well as 2) α2δ-1-independent mechanisms, including activation of KCNQ3/5 voltage-gated potassium channels (
7) and expression of δ-subunit-containing GABA
A receptors (
8). Each of these molecular mechanisms reduces excitatory/glutamatergic and/or enhances inhibitory/GABAergic neurotransmission.
Comparatively few studies have investigated the mechanisms of gabapentin’s effects on alcohol drinking and withdrawal in AUD. Because the neurobiological effects of drinking (i.e., inhibition of neuronal signaling via binding to GABA receptors and inhibiting glutamatergic synapses [
13,
14]) involve the same neurochemical mechanisms as gabapentin, the literature on gabapentin’s mechanisms cannot be easily applied to AUD. A seminal preclinical investigation of gabapentin demonstrated decreased anxiogenic effects of withdrawal and dependence-induced self-administration of alcohol, mediated by decreased GABAergic transmission in the central amygdala (
15). Subsequent studies have found that gabapentin 1) prevents excessive excitatory synaptogenesis by antagonizing the interaction of α2δ-1 with thrombospondins, upregulated after intermittent ethanol exposure (
16), and 2) normalizes alcohol-induced deficits in delta (1–4 Hz) power during slow-wave sleep (
17). Conversely, a recent
1H-MRS study found no association between parieto-occipital cortex GABA levels after ≥1 week of gabapentin treatment in a cross-sectional convenience sample of individuals with AUD in early recovery (
18).
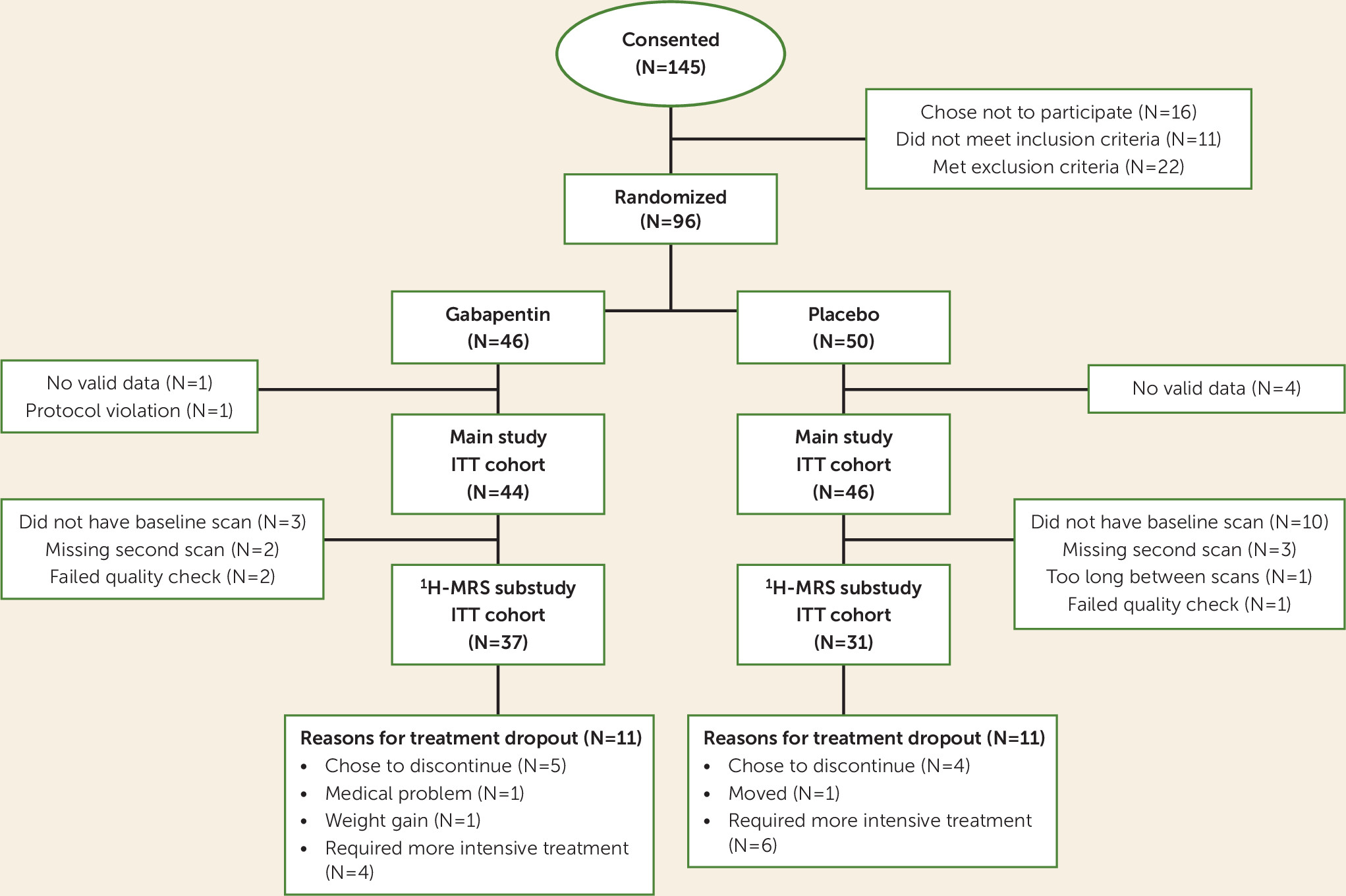
It is against this background that we report the results of the first controlled investigation of the neurobiological mechanisms of gabapentin treatment for AUD. In this MRI substudy of a previously reported clinical trial (
2), individuals with AUD, including a history of alcohol withdrawal syndrome and ≥72 hours of abstinence, were randomly assigned to receive gabapentin (1,200 mg/day) or placebo treatment for 16 weeks. Eligible participants additionally completed two
1H-MRS/MRI scans, one immediately preceding the first dose of study drug and the other after approximately 2 weeks of treatment. Here, we report the prospective effects of 1) early gabapentin treatment on GABA and glutamate levels in the dorsal anterior cingulate cortex (dACC), in the context of variable relapse drinking among participants, and 2) GABAergic and glutamatergic medication effects on further drinking across the remainder of the trial.
Discussion
Following recent demonstration of the antidrinking efficacy of gabapentin in individuals with AUD and a history of alcohol withdrawal syndrome (
2), the present analysis provides insight into the neurobiological mechanisms through which gabapentin may have worked to promote abstinence in that study. This preplanned investigation was based on the knowledge that adaptations in brain GABA and glutamate systems underlie AUD in general (
4) and alcohol withdrawal syndrome in particular (
23), and that potential gabapentin effects on those systems might explain its treatment efficacy (
15). Consistent with the preclinical and
1H-MRS gabapentin literature, gabapentin treatment was associated with significantly increased GABA levels and significantly decreased glutamate levels in the dACC, a fronto-cortical brain region. However, these associations were significantly moderated by (i.e., depended on) participants’ percentage of abstinent days during the first few weeks of treatment, which is not surprising given the large and dynamic impact that drinking itself has on brain glutamate and GABA levels (
14). Specifically, gabapentin (relative to placebo) was associated with 1) greater increases in glutamate and greater decreases in GABA levels in participants who remained mostly, or entirely, abstinent, but 2) greater decreases in glutamate and greater increases in GABA levels in participants who drank on more than approximately half of the days preceding the second scan. Furthermore, participants in the gabapentin group who had greater increases in glutamate (but not GABA) levels during the early weeks of treatment had significantly more percent days abstinent across the remaining study treatment period relative to those in the placebo group. Finally, lower baseline metabolite levels were associated with greater-magnitude metabolite-level changes across the first few weeks of treatment, and participants in the gabapentin group who had lower baseline glutamate levels had significantly more percent days abstinent across the study treatment period.
Consistent with the inhibitory effects of alcohol drinking on glutamatergic synapses, AUD has been consistently associated with reduced fronto-cortical glutamate levels in
1H-MRS studies (
38–
42), with the notable exception of during acute, clinically significant alcohol withdrawal, which has been associated with transient spikes in glutamate levels (
23). Although the present study focused on individuals with AUD and a history of alcohol withdrawal syndrome, those experiencing acute, clinically significant alcohol withdrawal were excluded from participation to ensure their safety (i.e., if they were receiving placebo). As a result, gabapentin-induced increases in dACC glutamate levels in the present study likely represented neurochemical shifts in the direction of abstinence-induced “normalization” rather than acute alcohol withdrawal effects, and this normalization depended on maintaining abstinence during the early treatment period. In contrast, a
1H-MRS investigation of acamprosate in individuals with AUD undergoing inpatient detoxification treatment for clinically significant alcohol withdrawal symptoms (with >60% of participants receiving benzodiazepines) found that acamprosate significantly decreased ACC glutamate levels relative to placebo, which was interpreted as a therapeutic “normalization” from the transient spike in glutamate levels that accompanies clinically significant alcohol withdrawal (
43). Unlike that study, however, we additionally demonstrated that medication-induced increases in glutamate prospectively predicted future abstinence.
Consistent with the facilitatory effects of alcohol drinking on GABAergic synapses,
1H-MRS-measured GABA levels were initially reported to be increased in individuals with AUD relative to healthy volunteers (
44), although subsequent studies failed to replicate this finding (
40,
45). The GABA findings from the present study could be viewed as consistent with results from that initial study, in that decreases in GABA levels were associated with increased percent days abstinent in participants in the gabapentin group, albeit only during the early treatment period. The GABA findings from the present study are also consistent with the seminal preclinical investigation of gabapentin for AUD, in which gabapentin was found to reduce excessive alcohol drinking by decreasing GABAergic transmission, thereby normalizing the alcohol-induced effect, in the central amygdala (
15).
Although the present study affirms
1H-MRS as a potentially valuable tool for exploring neurochemical drug effects, the interpretability of
1H-MRS findings is fundamentally limited by the methodology’s relatively low spatiotemporal resolution (
46,
47). Studies using
1H-MRS as a translational bridge (i.e., including both rodents and people) could overcome these limitations (
23) and provide more detailed molecular explanations for the gabapentin effects observed in people with AUD. Given the dynamic nature of neurochemical adaptation to alcohol drinking and withdrawal (
14,
48), more frequent
1H-MRS scanning during the initial days or weeks of gabapentin treatment could help disentangle the temporal interaction of changes in glutamate and GABA levels and percent days abstinent during the early phase of treatment. For example, consistent with findings from placebo-treated participants in the present study, we recently demonstrated that treatment-naive individuals with AUD had depleted dACC GABA levels, which normalized within 72 hours of monitored abstinence and which remained normal across a subsequent 5-day period of abstinence (
25). Although we found in the present study that both baseline and changes in dACC glutamate levels were associated with gabapentin’s promotion of abstinence, the findings involving changes in glutamate levels were nearly six times more statistically reliable than those involving baseline glutamate levels.
A notable strength of this study was that, unlike most
1H-MRS GABA studies (
49), we used a specialized MEGA-PRESS acquisition sequence that eliminated coedited “macromolecules,” which comprise 50% of the “GABA” signal (therefore denoted “GABA+”) acquired via traditional MEGA-PRESS acquisition (
29). Other strengths included a relatively large sample (e.g., more than twice that of Umhau et al. [
43]) and a relatively long duration of gabapentin treatment (i.e., 16 weeks, to address the often protracted nature of alcohol withdrawal symptoms [
50]), delivered in the context of a randomized double-blind placebo-controlled trial.
Limitations of the study include acquisition of 1H-MRS data from a single brain region, precluding conclusions concerning the regional specificity of findings; an MRI scanner upgrade that occurred partway through the study, although analyses demonstrated that results did not differ by scanner; exclusion of individuals with AUD who were experiencing acute, clinically significant alcohol withdrawal at the time of scanning; primary reliance on self-report alcohol consumption data; and inability to predict, at the individual level, who might respond best to gabapentin.
In sum, the results from this study suggest that gabapentin treatment promotes early abstinence partly by increasing dACC glutamate levels that are subsequently associated with gabapentin’s efficacy in reducing drinking over an extended period in individuals with AUD and a history of alcohol withdrawal syndrome. These novel findings contribute significantly to our understanding of how gabapentin may work to prevent relapse drinking in certain individuals with AUD who attempt abstinence, and are consistent with our previous report of gabapentin’s efficacy (
2). They also provide evidence for a biomarker of efficacious treatment (i.e., increased dACC glutamate levels) that may be used to evaluate other glutamatergic and/or GABAergic medications for individuals with AUD, and potentially other conditions marked by dACC glutamate and/or GABA deficiency (e.g., cannabis use disorder [
51] and co-occurring bipolar disorder and substance use disorder [
52]).