Neurodegenerative diseases (NDDs) are common and impose substantial morbidities and costs on patients, caregivers, and society.
1 The two most common, Alzheimer's disease (AD) and Parkinson's disease (PD), affect more than 5 million people in the United States and are exponentially increasing with population demographic trends;
2 and they result in considerable morbidities and costs.
3 Neuropsychiatric disturbances are prominent among these morbidities and can contribute significantly to quality of life in NDDs.
1 Clinicians prescribe psychotropic drugs to treat the neuropsychiatric disturbances in NDDs
3 without regard to their potential neuroprotective effects. Psychotropics affect the intracellular mechanisms that are common to the pathobiology of a variety of NDDs (i.e., pathological proteins, proteasome, mitochondria, apoptosis), suggesting the possibility that psychotropics may function neuroprotectively across a number of these diseases.
1,3 The potential to modify the course of a disease through these effects has substantial implications for both patients and society.
1,4 The neuroprotective effects of established psychotropic medications are worth investigating, given the unsatisfying results from clinical trials of newer neuroprotective agents. Also, there is considerable advantage to re-purposing established psychotropic drugs because much is already known about their absorption, metabolism, excretion, toxicity, safety, blood–brain barrier penetration.
This report updates the literature of FDA-approved first-line psychotropics to June 10, 2010. Over the intervening 33 months, there has been an exponential increase in relevant publications. We evaluated these data according to the previously-published provisional criteria,
1,3 shown in
Table 1. This report focuses on common neurodegenerative intracellular effects in mature neural tissues. Neurons and glia may behave differently from non-neural cells, and immature and mature neurons can behave differently,
6 and even, sometimes, oppositely
7 with respect to apoptosis. For example, neurons and SK-N-SH neuroblastoma cells are differentially sensitive to antipsychotic-induced apoptosis.
6 Since NDDs generally involve mature neurons and glia, we therefore confined the present literature review to these mature cells and excluded studies done in cell-lines related to neuroblastomas (e.g., SK-N-SH, Neuro-2a, N1E-115, etc.), pheochromocytomas (e.g., PC12, KNA, KAT45, etc.), other malignancies, and other immature cell-lines. For this reason, assessment by
Table 1 preclinical criteria is limited to findings in mature neural tissues.
Pending criteria predictive validity, the findings below represent an advance and refinement in selecting currently-prescribed psychotropics as candidates for disease-modifying clinical trials in NDDs.
METHOD
Preclinical findings in mature neural tissue and clinical findings in NDDs treated with psychotropics were evaluated by
Table 1 criteria. Relevant studies were identified through a literature search (PubMed search terms, February 1, 2010: (alpha-synuclein OR beta-amyloid OR tau OR TDP-43 OR ubiquitin OR proteasome OR mitochondrial viability OR mitochondria OR mitochondrial transition pore OR cytochrome c release OR endosome OR lysosome OR autophagy OR endoplasmic reticulum OR leukocyte viability OR apoptosis) AND (pramipexole OR ropinirole OR amantadine OR haloperidol OR fluphenazine OR trifluoperazine OR thiothixene OR chlorpromazine OR thioridazine OR risperidone OR olanzapine OR quetiapine OR ziprasidone OR aripiprazole OR clozapine OR paliperidone OR iloperidone OR asenapine OR tetrabenazine OR pimavanserin OR lithium OR carbamazepine OR oxcarbazepine OR valproate OR amitriptyline OR imipramine OR nortriptyline OR desipramine OR clomipramine OR trimipramine OR doxepin OR protriptyline OR maprotiline OR bupropion OR fluoxetine OR sertraline OR fluvoxamine OR paroxetine OR citalopram OR s-citalopram OR trazodone OR nefazodone OR venlafaxine OR duloxetine OR mirtazapine OR buspirone OR diazepam OR chlordiazepoxide OR flurazepam OR temazepam OR chlorazepate OR clonazepam OR lorazepam OR oxazepam OR alprazolam OR zaleplon OR zolpidem OR zopiclone OR s-zopiclone OR cyproheptadine OR hydroxyzine OR benztropine OR trihexyphenidyl OR modafinil OR melatonin OR ramelteon) AND (neuron OR neuronal OR neurons OR glia OR glial OR neuroglia)).
Citations were reviewed to exclude findings in non-neural tissue and immature or malignancy-related cell lines. The review was limited to mature neural tissues (except in disease-specific animal models, where stem cells in mature brain were also considered) for reasons detailed above. We considered studies of any methodology but excluded models distinct from NDD (e.g., stroke, tardive dyskinesia, etc.). Cell culture conditions not typical of NDD (e.g., hyperosmotic stress, oxygen deprivation, etc.) were also excluded. The sole exception involved mature cerebellar granule cells (CGNs) that require hyperkalemic conditions for survival and reliably undergo apoptosis after withdrawal of serum or potassium.
8We focused on intracellular processes common across NDDs specified in the search terms (proteins, ubiquitin–proteasomal system, mitochondria, and apoptosis), and did not consider studies of intracellular calcium influx or other disease mechanisms unless those studies also considered the intracellular processes of interest. DNA fragmentation and condensation was required to ascertain apoptosis, whereas other indices (apoptotic mediator concentration, cell viability) were considered insufficient. The single exception to this involved CGN studies (as noted, CGNs are well documented to undergo apoptosis with serum/potassium deprivation). A single reviewer evaluated each abstract to determine whether it might potentially provide information on relevant intracellular processes after meeting the above methodological inclusions, even if not clearly stated, in which case the article itself was then reviewed.
In assessing Preclinical Criterion 6 (disease-specific animal models), outcomes consistent with putative neuroprotection were considered even if the study did not specifically address the intracellular targets required for cell and tissue studies, provided that outcomes were relevant to disease-specific clinical outcomes. In interpreting the results and applying the findings in specific NDDs, findings from mature neural tissues in a disease-specific animal model were considered to be relevant to only that disease, whereas data pertaining to neural stem-cells in mature animals were considered to be potentially applicable to any disease, regardless of model.
Two studies considered combined treatment of lithium with valproate. The results of this combined treatment were considered separately and not included in the neuroprotective scores for either drug unless findings pertain to either drug given by itself.
Clinical findings evaluated by
Table 1 Clinical Criteria were identified through a literature search (PubMed search terms, June 10, 2010: (pramipexole OR ropinirole OR amantadine OR haloperidol OR fluphenazine OR trifluoperazine OR thiothixene OR chlorpromazine OR thioridazine OR risperidone OR olanzapine OR quetiapine OR ziprasidone OR aripiprazole OR clozapine OR paliperidone OR iloperidone OR asenapine OR tetrabenazine OR pimavanserin OR lithium OR carbamazepine OR oxcarbazepine OR valproate OR amitriptyline OR imipramine OR nortriptyline OR desipramine OR clomipramine OR trimipramine OR doxepin OR protriptyline OR maprotiline OR bupropion OR fluoxetine OR sertraline OR fluvoxamine OR paroxetine OR citalopram OR s-citalopram OR trazodone OR nefazodone OR venlafaxine OR duloxetine OR mirtazapine OR buspirone OR diazepam OR chlordiazepoxide OR flurazepam OR temazepam OR chlorazepate OR clonazepam OR lorazepam OR oxazepam OR alprazolam OR zaleplon OR zolpidem OR zopiclone OR s-zopiclone OR cyproheptadine OR hydroxyzine OR benztropine OR trihexyphenidyl OR modafinil OR ramelteon) AND (clinical trial OR randomized controlled trial) AND (neuroprotection OR neuroprotective OR disease-modifying OR disease modifying OR disease modification OR progression OR disease progression OR biomarker OR beta-amyloid OR tau protein OR alpha-synuclein OR huntingtin OR TAR DNA binding protein 43 OR TAR DNA-binding protein 43 OR TDP-43 OR TDP 43 cerebrospinal fluid OR imaging OR magnetic resonance imaging OR single photon emission computed tomography OR positron emission tomography) AND (neurodegenerative disease OR Alzheimer's disease OR Parkinson's disease OR Huntington's disease OR frontotemporal lobar degeneration OR FTLD OR FTD OR frontotemporal dementia OR ALS OR amyotrophic lateral sclerosis OR progressive supranuclear palsy OR corticobasal degeneration OR Pick's disease OR MSA OR multiple system atrophy)). Methods specific to the quantitative evaluation of
Table 1 criteria are described in respective Results sections.
DISCUSSION
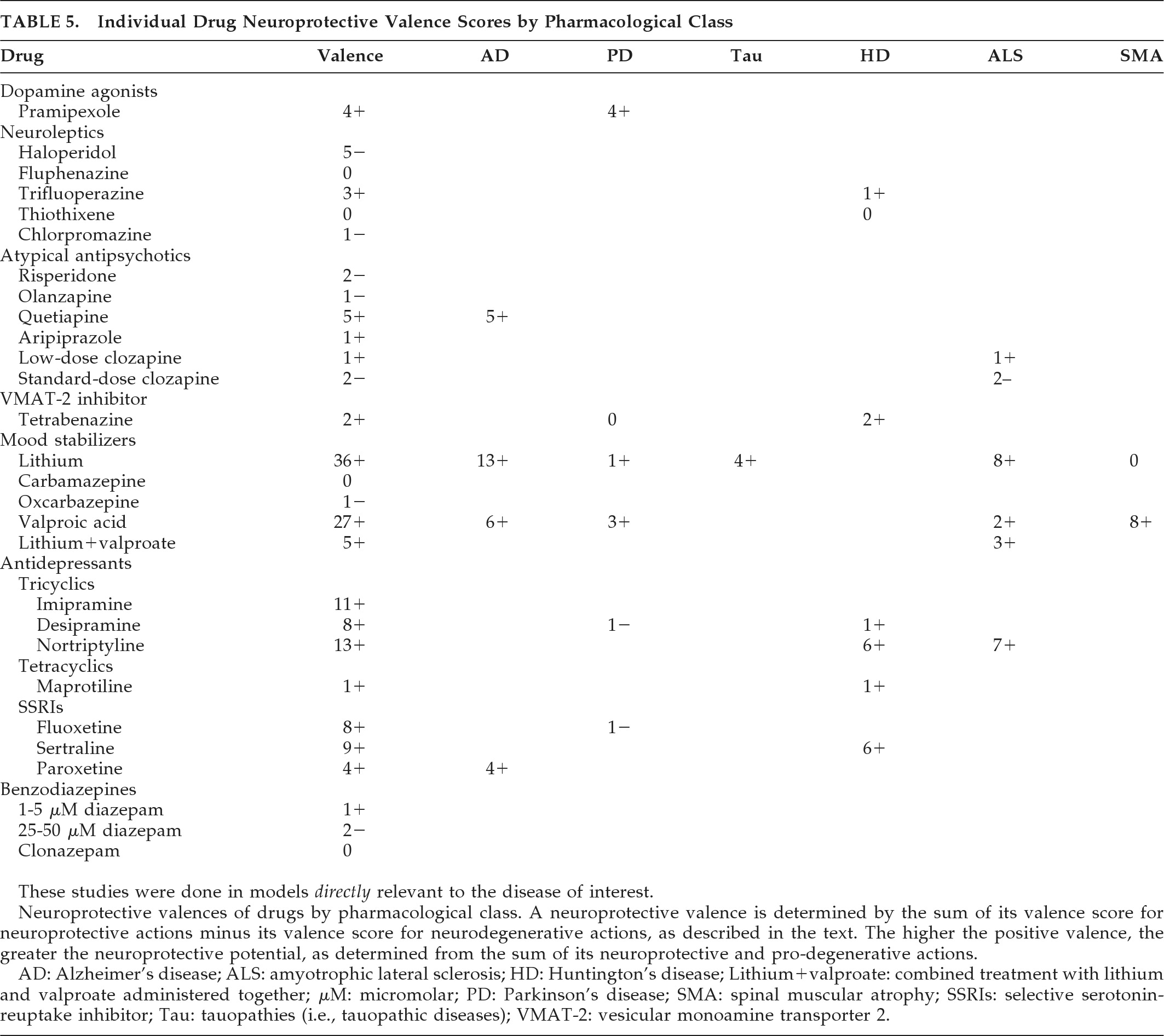
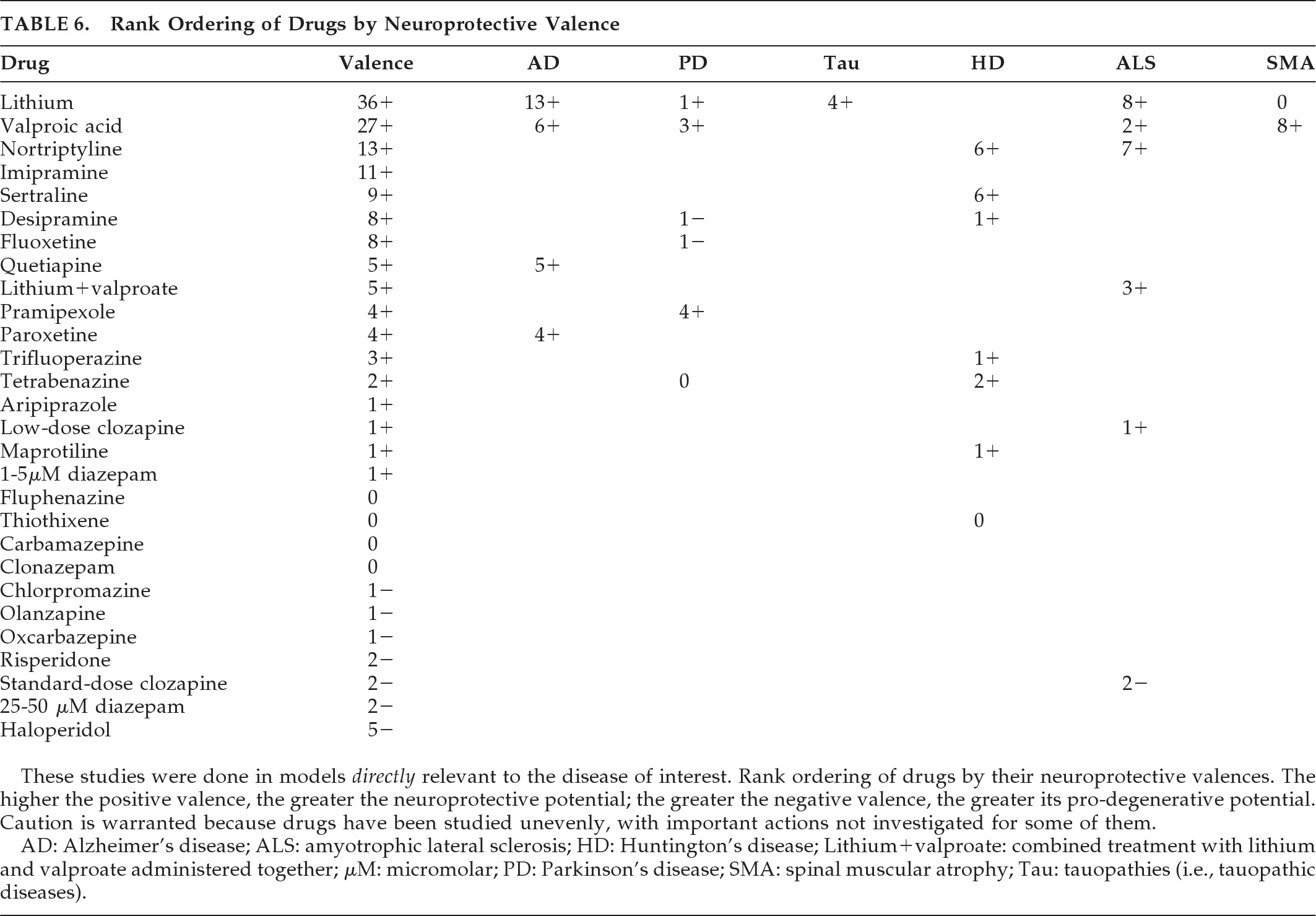
This report applies a rigorous inclusion requirement and a presumptively valid quantitative method to produce a broad comprehensive review of commonly-prescribed psychotropic agents that could double as neuroprotective therapies. The findings indicate a potential neuroprotective role for lithium, paroxetine, or lithium-plus-valproate in AD, lithium in frontotemporal lobar degeneration (FTLD) and in combination with valproate in ALS, pramipexole and valproate in PD, amantadine and buspirone in MSA, and antidepressants in HD. This report, limited to psychotropic medicines, should prove of special relevance to neuropsychiatrists and others who use these drugs in NDD with neuropsychiatric symptoms.
The best-documented, most robust, and promising preclinical findings are shown in
Table 7 and are considered to have the highest probability of translating to clinical neuroprotection in human patients. The neuroprotective index scores of this table reflect replicated findings in drugs with proven benefit in disease-specific animal models and a net positive neuroprotective valence. Dopamine agonist, neuroleptic, atypical antipsychotic, and the VMAT-2 inhibitor tetrabenazine findings did not fulfill the
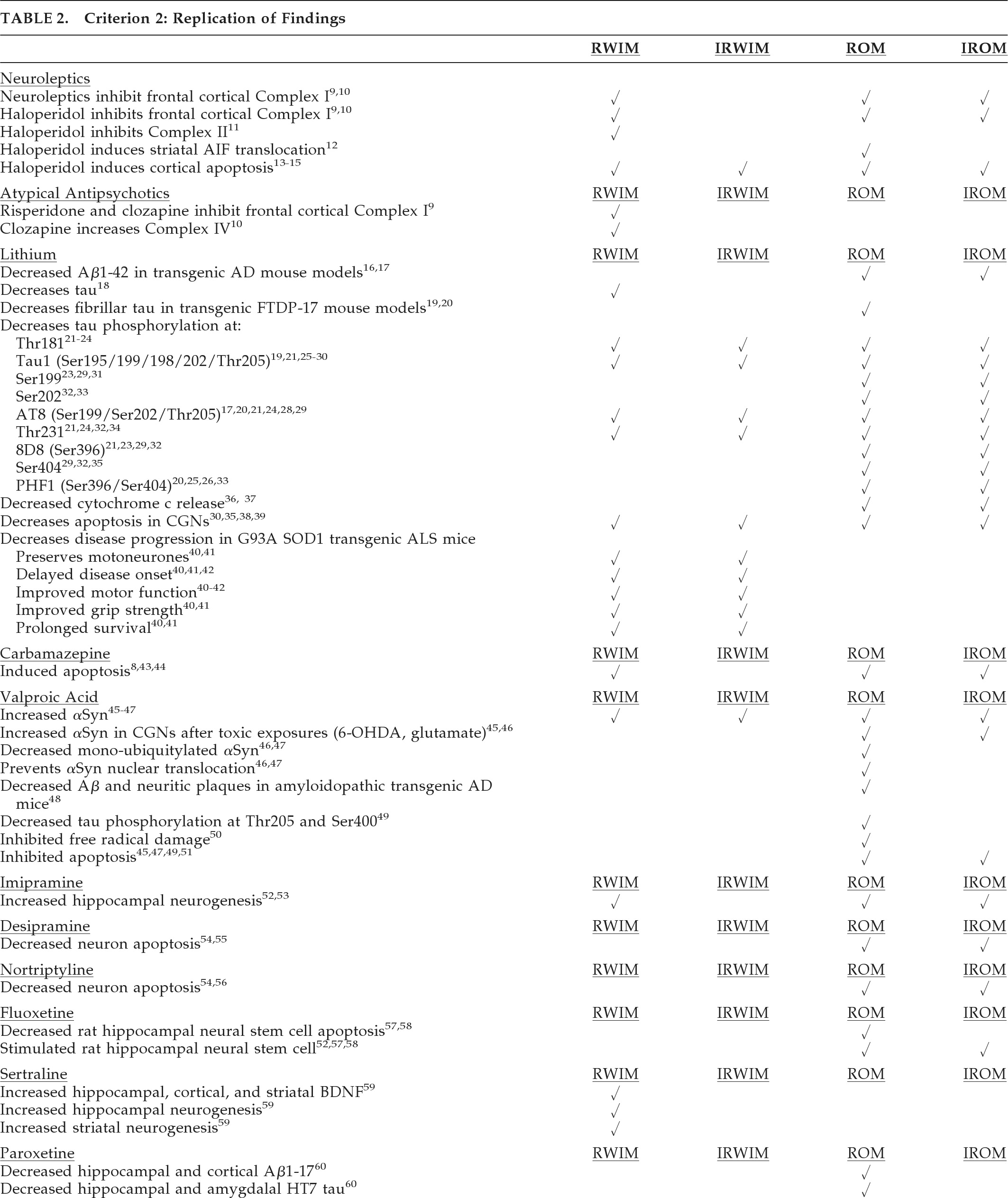
Table 7 composite criteria. Lithium showed potential neuroprotection in reducing Aβ1–42, tau phosphorylation, and fibrillar tau in aging, amyloidopathic, and tauopathic mouse models (
Table 7). In particular, tau phosphorylation was reduced at epitopes relevant to AD (Thr181, Ser199, AT8, Thr231, Ser396) and tauopathies including PSP, FTLD, and FTDP-17 (Tau1, Ser202, AT8, Thr231, Ser396, Ser404), and MSA (Tau1).
In the G93A SOD1 transgenic mouse model of ALS, lithium showed both neuronal and overall neuroprotective properties in most studies. Valproate reduced Aβ and neuritic plaques in amyloidopathic transgenic mouse models of AD and increased αSyn while decreasing nuclear translocation in the rotenone rat model of PD. The antidepressant paroxetine reduced cortical and hippocampal Aβ processing and hippocampal HT7 human tau in a transgenic model of AD. In HD mouse models, nortriptyline and desipramine preserved caudate medium spiny neurons against glutamate apoptosis, and sertraline increased both BDNF and hippocampal and striatal neurogenesis. Thus, the most robust preclinical findings detailed in
Table 7 suggest neuroprotective potential for lithium, valproate, and paroxetine in AD, lithium in tauopathies and ALS, valproate in PD and perhaps other synucleinopathies, and several antidepressants in HD.
Although a number of psychotropics have been studied in AD, PD, MSA, and ALS, the only conclusions that can be drawn from these clinical trials are that olanzapine may deteriorate cognition in AD, and lithium is probably not a useful neuroprotectant in most ALS cases, at least when given with riluzole. Initial findings needing replication include lithium neuroprotection in AD with increased BDNF levels and in
early ALS, and the futility of r-pramipexole and valproate in ALS. Neuroprotective paradigms are needed to resolve the neuroprotective efficacy of ropinirole and pramipexole in PD, and amantadine and buspirone (using more sensitive measures) in MSA. Clinical studies have relied on a difference in slope-deterioration between treatments to measure progression rather than delayed-start or randomized-withdrawal designs.
5 At present, no disease-modifying neuroprotective agents have been conclusively demonstrated in clinical trials.
Additional studies in the literature require more detailed discussion. Moderate neuroprotective index scores for motor-neuron survival and motor function with lithium in the ALS G93A SOD1 mutation mouse model (
Table 7; scores of 32 and 16, respectively, and an average of 14.4 across ALS findings, out of possible scores of 1 to 64) contrast somewhat with negative clinical findings, although it has been argued that this mutation model is not representative of human disease, and the mutation is present only in a small minority of ALS patients.
Moderate scores in AD models with tau mutations suggest therapeutic potential for lithium in tauopathic diseases, although poor tolerability resulted in premature discontinuation in 13 of 14 patients with progressive supranuclear palsy (PSP) or corticobasal degeneration (CBD) during a 28-week, open-label, safety and tolerability trial (recently published as an abstract).
105 For valproate, a low-to-moderate neuroprotective index score was achieved for Aβ neuritic plaque reduction in AD amyloid- and presenilin-mutant mice. Forthcoming results of a 26-month, randomized, placebo-controlled trial in 313 patients with AD with clinical rating, biomarker, and brain-volume outcomes
106 (manuscript in review process) indicate no difference in slopes of deterioration on clinical ratings (Tariot PN, personal communication; January 2011). The valproate group showed greater loss in hippocampal and whole-brain volume, accompanied by greater ventricular expansion. It is unclear whether these changes may be due to direct drug effects such as osmotic shifts or metabolic toxicity or influences on AD pathology, and it is unknown whether these effects are reversible or clinically relevant. Although valproate levels averaged 40–50 μg/ml, the levels may not have been sufficient to engage molecular targets (Tariot PN, personal communication). Alternatively, these negative results may reflect a translational predictive failure of Aβ neuritic plaque or amyloid transgenic mouse models. After all, the neuroprotective valence and neuroprotective index score are only as good as the models that they are based upon, and model and tolerability confounds prevent a current assessment of their predictive capability. It is also possible that higher and more robust neuroprotective index scores are needed to predict clinical neuroprotection, since even moderate scores were not consistently obtained across the findings and models of
Table 7. Toward this, simultaneous administration of lithium-plus-valproate might potentially produce quantitatively greater and more robust neuroprotective index scores and result in clinical therapeutic effects.
There are several caveats in interpreting how well the present findings generalize and can predict clinical translation. First, we focused on findings in mature neural tissues because of substantial differences in how mature and immature tissues behave with respect to the common intracellular processes considered here, yet it remains possible that some potentially relevant preclinical findings might be excluded by this requirement. Second, the concepts of neuroprotective valence and the composite neuroprotective index score remain to be validated. Although it makes sense that drugs with a multiplicity of different neuroprotective actions and minimal prodegenerative effects (i.e., higher positive valence) will more likely be neuroprotective in human clinical trials, this concept remains to be validated, especially in light of disappointing results for lithium, a drug with a high valence, in ALS. Forthcoming data will also indicate whether the neuroprotective index score is a predictor of clinical neuroprotection in humans.
There are the limitations inherent to a review of the literature. Although the best strategy available, the literature search may have missed some data because of indexing inconsistencies. Several studies were not identified by the preclinical
42,69,95,99 and clinical
78,80 search strategies, but rather by bibliographic extension. Furthermore, the search strategy targeted the proteins, organelles, and processes specified, and although this produced a number of citations pertaining to additional neurodegenerative processes, the review cannot be considered to be fully comprehensive with respect to processes such as “oxidation.” Obviously, this review also does not consider additional neuroprotective mechanisms, for example, “inflammation.” There are also reporting biases in the literature. Drug actions have been investigated unevenly and unsystematically across and within neuroprotective actions, and animal models vary in their translational predictive power.
There are several perspective-related issues that can make the interpretation of some of the results difficult at this point. For example, an increase in αSyn can lead to proteasomal inhibition, apoptosis, and inclusion formations including Lewy bodies, pathological tau, and Aβ and yet a rise in αSyn can also effect a neuroprotective response. Proteasomal inhibition would seem to be pro-apoptotic, and yet the administration of subtoxic proteasome inhibitors has been associated with neuroprotection.
100 In the case of αSyn, mono-ubiquitylation and nuclear translocation predict apoptosis, in contrast to a non-ubiquitylated rise in αSyn concentration.
47 Nevertheless, seemingly similar responses can, at times, signal
either neuroprotection or neurodegeneration, and whether a particular response is neuroprotective or proapoptotic may also depend on both its extent and whether it is occurring early or late in the disease process.
There are several drug-related issues that can affect generalizability and translational prediction. Drug dose can make a difference in effects. Low doses of lithium, for example, activate autophagy in extraneuronal models,
101 whereas these same doses may be ineffective in activating other neuroprotective functions. On the other hand, in preclinical models of neuroprotection, a bell-shaped rather than open-ended sigmoid-shaped dose-response curve is often encountered. Thus, too high a dose may exceed the therapeutic window of neuroprotection for some mechanisms. This could help explain the diverging effects of low versus standard doses of clozapine
69 and diazepam.
61,102 Results may also vary with the type of apoptogen, neurotoxin, and locus of action. For example, valproate prevented nigral loss in the rotenone,
47 but not the MPTP
103 mouse model of PD. Some neuroprotective actions of psychotropics may relate to their pharmacodynamic mechanisms.
3 For example, 5HT
1a receptor agonists may inhibit apoptosis through nerve growth factor signaling.
104 Duration of treatment can affect neuroprotection as well. For example, hippocampal Bcl-2 expression is upregulated by chronic but not acute treatment with antidepressants.
58,64 Taken together, a given psychotropic can have plural neuroprotective mechanisms and, simultaneously, have pro-degenerative effects, with the capacity to act as both friend and foe, depending on dosing and context.
In conclusion, despite limitations and other issues in interpreting the literature, several established psychotropic medications have sufficient promise to warrant further study as neuroprotective agents. We need clinical trials using designs adequate to assess disease-modification
5,107 as follow-up to these preclinical and clinical data. Priorities include the agents identified by the provisional preclinical neuroprotective criteria and resolution of initial findings in the clinical trials, including dopamine agonists (PD, ALS), amantadine (MSA), lithium (AD with elevated BDNF, tauopathies, early ALS), valproate (AD, PD, synucleinopathies, ALS), lithium-plus-valproate (AD, ALS), nortriptyline (HD), desipramine (HD), sertraline (HD), paroxetine (AD), and buspirone (MSA-C). Secondary priorities include study of the other agents identified by each of the provisional preclinical neuroprotective criteria in isolation. It will be of interest to identify which provisional criteria prove to be most predictive of clinical translational benefit. Equivocal results in clinical neuroprotective trials will signal the need to develop better preclinical models with improved abilities to predict translational treatments in human disease.