Inadequate response to treatment in a substantial proportion of affected individuals contributes to the large burden of disability associated with major depressive disorder (
1). It has been proposed that outcomes of depression could be improved by personalizing treatment with the use of biomarkers that are easily measurable by noninvasive methods and are differentially predictive of outcomes with alternative treatments, with clinically significant effect sizes (
2). While replicable general predictors of poor treatment outcomes have been identified (
3,
4), clinically significant differential predictors are lacking.
Several lines of evidence implicate immunity and inflammation in the pathogenesis of depression and mechanisms of antidepressant response (
5). Increased levels of systemic inflammation have been associated with depression (
6). Inflammatory biomarkers have also been associated with an adverse course of depression at the population level (
7). Different effects of norepinephrine and serotonin on inflammation suggest that inflammatory biomarkers may differentially predict outcomes of treatment with antidepressants that affect the levels of these neuromediators (
8). Norepinephrine inhibits production of Th1 proinflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), by white blood cells and microglia through their action on the β
2 receptors (
8–
10). Serotonin inhibits the production of Th2 cytokines such as interleukin 6 (IL-6) (
8). Antidepressant drugs that affect norepinephrine and serotonin also distinctly affect immunity: while norepinephrine reuptake inhibitor antidepressants suppress Th1-type cytokines and shift the balance toward humoral immunity, serotonin reuptake inhibitors reduce the production of Th2-type cytokines and shift the balance toward cellular immune response (
8,
11). Nortriptyline, a tricyclic antidepressant whose predominant mode of action is inhibition of norepinephrine reuptake, has also been found to inhibit migration of polymorphonuclear white blood cells toward an inflammation site, an effect not replicated with serotonin reuptake inhibitors (
12). Inflammatory cytokines also affect the metabolism of serotonin and norepinephrine in different ways: while TNF-α increases the expression of serotonin transporter by astrocytes, potentially reducing the levels of intracellular brain serotonin (
13), interleukin 1 (IL-1), another Th1 proinflammatory cytokine, increases the production of norepinephrine in the hypothalamus (
14). These data led us to formulate the hypothesis that pretreatment levels of systemic inflammation will differentially predict therapeutic response to the serotonin reuptake inhibitor escitalopram and the norepinephrine reuptake inhibitor nortriptyline in major depressive disorder. While previous studies have shown that baseline levels of inflammatory biomarkers predicted response to a single treatment (
15–
17) and were related to overall resistance to antidepressants (
18,
19) or persistence of depression among antidepressant-treated individuals (
7), to our knowledge no previous study has examined inflammatory biomarkers as differential predictors of response to alternative treatments.
A number of proteins in peripheral blood can be used as markers of systemic inflammation. We chose C-reactive protein (CRP), an acute-phase protein produced by the liver in response to interleukin 1β (IL-1β) (
20). The choice was based on pragmatic considerations. High-sensitivity measurement of CRP is a readily available test in most medical biochemistry laboratories, the level of CRP does not change with time of day or time since last meal, and CRP is stable in stored biological samples (
21,
22). Of all evaluated inflammatory biomarkers, CRP has the largest body of evidence for consistent associations with depression, its risk factors, and its treatment outcomes (
6,
16,
23). Therefore, we specified and tested the practical hypothesis that CRP level will differentially predict response to escitalopram and nortriptyline in depression.
Discussion
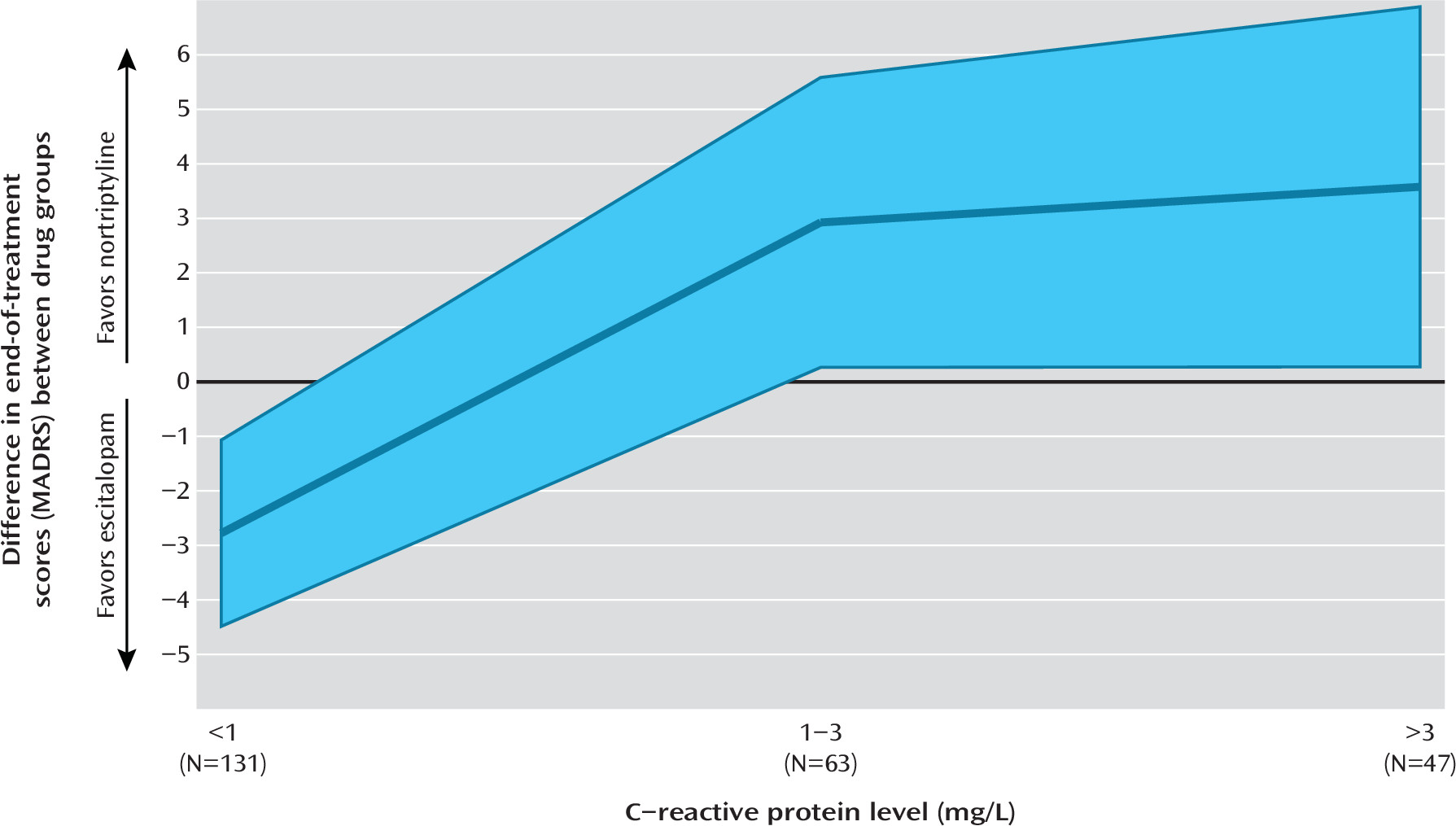
In a study of 241 patients with depression, we found that CRP, an easily accessible biomarker of systemic inflammation, differentially predicted response to escitalopram and nortriptyline. The effect size of the prediction suggested that the use of this biomarker in the selection of an antidepressant may meaningfully improve outcomes in the treatment of depression.
A major advantage of CRP lies in its easy availability. CRP can be obtained from a nonfasting peripheral blood sample, and high-sensitivity assays are routine in most medical laboratories (
22). In GENDEP, CRP differentially predicted therapeutic response to the two antidepressants, suggesting that it could be used to inform the selection of antidepressant drug (
2). The effect size of the differential prediction met criteria for clinical significance (
43,
44), suggesting that the prediction can be meaningful in individual cases. The even stronger effects with the secondary self-report outcome measure (BDI) suggests that the differential prediction will be as meaningful to patients as to clinicians. Additivity with other predictors, such as interest-activity symptoms (
4), suggests that an even more accurate prediction of outcome can be achieved by the combination of biomarkers and additional clinical variables (
3). While the prediction of outcome by interest-activity symptoms replicated across studies (
4), the contribution of CRP level and the effect size of the joint prediction will require replication to establish generalizability. This promising finding contrasts with the limited effects of pharmacogenetics (
48,
49) and suggests that state-dependent biomarkers may be needed to personalize treatment choice (
50). The added value of combining state-dependent biomarkers with genetic ones is a field for future investigation.
While this is the first study that shows a differential prediction by an inflammatory biomarker of response to serotonin reuptake inhibitor and norepinephrine reuptake inhibitor antidepressants, the results are consistent with the literature. Two previous studies (
17,
19) found that higher levels of CRP or proinflammatory cytokines at baseline were associated with poorer response to the predominantly serotonin reuptake inhibiting antidepressants, including escitalopram, fluoxetine, and low-dosage venlafaxine. Although one study (
51) found no significant effect of baseline inflammation on the therapeutic response to sertraline in individuals with cardiovascular disease, the mean CRP levels in that selected sample were four times those in the present study, putting most subjects in the high-risk range of CRP levels and making the data sets incomparable. In previous studies using nortriptyline or related tricyclic antidepressants, elevated CRP level either was not associated with outcome or predicted better antidepressant response, consistent with the present findings (
15,
52). Baseline CRP levels were also found to moderate the antidepressant effects of a treatment that directly targets immune pathogenic mechanisms, which was beneficial only for individuals with high CRP levels (
16). Additional hope for individuals with high levels of systemic inflammation comes from a study that shows that they may benefit more from physical exercise (
53). Taken together, the evidence to date is consistent with systematic inflammation acting as a moderator of antidepressant response.
The hypothesis that led to this investigation stemmed from known contrasting effects of norepinephrine and serotonin on the immune system and hence focused on the differential action of norepinephrine reuptake inhibitor and serotonin reuptake inhibitor antidepressants (
8–
10,
12). However, the differential prediction of response to these types of antidepressants is currently based on a single study of two drugs and must be interpreted with several limitations kept in mind.
First, the two antidepressants investigated here differ in a number of respects. Escitalopram is a highly selective serotonin reuptake inhibitor with no important effects on other receptors or transporters and no effect on the production of proinflammatory cytokines (
26,
54). Nortriptyline is a tricyclic antidepressant; although it preferentially blocks the reuptake of norepinephrine and has only a weak affinity to the serotonin transporter (
27), it also binds several receptors, including the histamine H
1 receptor, the muscarinic acetylcholine receptor M
1, and serotonin receptors 5-HT
2A and 5-HT
2C (
55). Given the pleiotropy of nortriptyline effects, the present study cannot separate the effects of norepinephrine reuptake blockage from other compound-specific effects or class effects of tricyclic antidepressants. It remains to be investigated whether other antidepressants with similar effects on the immune system (
8) can substitute for nortriptyline in patients with high levels of systemic inflammation.
Second, this study is limited to testing a pragmatic hypothesis regarding prediction of treatment outcomes. In the absence of serial measurements of multiple cytokines, it does not directly address the molecular mechanisms underlying the observed effects. We are unable to differentiate between effects of antidepressants on cytokine production (
8) and effects of cytokines on the monoaminergic neurotransmitter systems affecting the therapeutic action of antidepressants (
13). Studies with serial collection of samples over time are needed to elucidate the molecular mechanisms underlying the observed effect.
Third, GENDEP was primarily a pharmacogenetic study (
56,
57), and collection of serum samples was not a priority. As a result, serum samples were available for just over half of the participants. Participants with and without serum samples were comparable on depression severity, drug allocation, and treatment outcome, suggesting no selection bias. However, the smaller sample size may reduce the generalizability of findings in spite of high levels of statistical significance. Replication is a necessary step before clinical implementation. Future studies should test the replicability of the differential prediction of escitalopram and nortriptyline efficacy by CRP level and extend the findings to other antidepressants and other markers of inflammation.