Abnormalities in the neural circuitry of the dorsolateral prefrontal cortex may contribute to disturbances in certain cognitive functions, such as working memory, that are characteristic of schizophrenia
(1,
2). Convergent lines of evidence suggest that these circuitry abnormalities involve alterations in the inhibitory neurotransmitter γ-aminobutyric acid (GABA). For example, the activity of glutamic acid decarboxylase (GAD), the enzyme responsible for GABA synthesis, and the level of expression of GAD
67 mRNA have both been reported to be reduced in the dorsolateral prefrontal cortex of subjects with schizophrenia
(3–
6). In addition, alterations in GABA
A receptors have been reported in ligand binding studies
(7).
These changes in markers of GABA neurotransmission may reflect abnormalities that are selective for the chandelier subclass of GABA-containing neurons
(8). The axon terminals of chandelier neurons form distinctive arrays, termed “cartridges,” that provide inhibitory input exclusively to the axon initial segment of pyramidal cells
(9). Because of the location of the synapses formed by their axon terminals, chandelier cells are positioned to powerfully regulate the output of pyramidal neurons, the principal type of cortical excitatory projection neuron
(10). We recently reported
(8) that the density of these axon cartridges, as revealed by their immunoreactivity for the GABA membrane transporter (GAT-1), was decreased on average by 40% in the dorsolateral prefrontal cortex of schizophrenic subjects. In addition, these abnormalities may be specific to the chandelier class of GABA neurons, since the axon terminals of other classes of GABA neurons did not appear to be affected
(8). Given the critical role of chandelier cells in dorsolateral prefrontal cortex circuitry, alterations in GAT-1-immunoreactive axon cartridges could play a major role in the dysfunction of this brain region in schizophrenia.
To further explore the pathophysiological significance of alterations in chandelier neuron axon cartridges, we asked the following questions. First, does the decreased density of GAT-1-immunoreactive cartridges reflect a change in inhibitory control over all populations of pyramidal cells in the dorsolateral prefrontal cortex, or are certain subpopulations preferentially affected? Since pyramidal cells in each cortical layer receive specific types of afferent inputs and furnish excitatory projections to certain brain regions
(11), determining whether chandelier cell axon cartridges are differentially affected across cortical layers will provide insight into the integrity of specific dorsolateral prefrontal cortex circuits in schizophrenia. Second, does the decrease in density of GAT-1-immunoreactive cartridges in schizophrenia reflect an effect of antipsychotic or other medications commonly used in the treatment of schizophrenia? The answer to this question will help reveal the pathophysiological versus the therapeutic significance of alterations in chandelier neuron axon cartridges.
To address these questions, we determined the laminar distribution of GAT-1-labeled chandelier neuron axon cartridges in the dorsolateral prefrontal cortex of 30 schizophrenic and 60 comparison subjects. In addition, we used an identical approach to examine GAT-1-immunoreactive axon cartridges in nonhuman primates chronically treated with haloperidol and benztropine in a fashion that mimicked clinical use.
METHOD
Specimens from 90 human brains (
table 1) were obtained, during autopsies conducted at the Allegheny County, Pa., coroner’s office, with the consent of next of kin and the approval of the Health Sciences Institutional Review Board of the University of Pittsburgh. Gross and microscopic neuropathological screening revealed no abnormalities in any subject, with the following exceptions. Thioflavine S staining revealed a rare neurofibrillary tangle in one subject (number 493) and a few neuritic plaques in four subjects (numbers 207, 213, 304, and 313). However, the density of the plaques was insufficient to meet the diagnostic criteria for Alzheimer’s disease
(12), and none of the subjects had a history of dementia. For two subjects, the cause of death involved brain damage (number 207: left parietal subdural hematoma; number 517: vascular malformation and intracerebral hemorrhage in the right temporal lobe), but the area of interest for the present study was not affected. An independent panel of three experienced clinicians made consensus DSM-III-R diagnoses, using information obtained from medical records and structured interviews with surviving family members. These procedures have been previously described
(13).
We examined 30 subjects with schizophrenia, each of whom was individually matched to one normal and one nonschizophrenic psychiatric comparison subject on age, sex, and postmortem interval (
table 1). There were 10 female and 20 male subjects in each of the three groups of subjects. The postmortem interval was defined as the time between the estimated time of death and the time when the brain was placed in ice-cold 4% paraformaldehyde. Individual subject matching on these variables was done to minimize their potential confounding effects on our outcome measures. Mean values for age, postmortem interval, and percentage of out-of-hospital deaths did not differ across subject groups (
table 2). Subjects with schizophrenia had an earlier age at onset of illness and a longer duration of illness than the psychiatric comparison subjects, and 14 psychiatric subjects had committed suicide as opposed to only six schizophrenic subjects (
table 2). In contrast, the schizophrenic and psychiatric subjects did not differ with regard to the incidence of comorbid alcohol or other substance disorders (
table 2). One of the schizophrenic subjects (number 234) had never been treated with antipsychotic medications, and six others had been off these medications for a period of time ranging from 1 month to 10 years prior to death (
table 1). Eight of the psychiatric subjects had been treated with antipsychotic medications at sometime during their lives (
table 1). The first 15 subjects in each diagnostic group (i.e., triads 1–15 in
table 1) had been used in our previous study of GAT-1-immunoreactive cartridges
(8).
Tissue Preparation and Immunocytochemistry
At autopsy, the brain was removed and the left hemisphere was cut into coronal blocks (1.0 cm thick). Tissue blocks were immersed in ice-cold 4% paraformaldehyde in phosphate buffer for 48 hours, washed in a graded series of sucrose solutions, and then stored in an antifreeze solution at –30°C. Tissue storage time did not differ across the three subject groups (
table 2). Previous studies
(13,
14) have shown that these storage conditions do not affect immunoreactivity.
Coronal sections (40 µm) were cut on a cryostat, and every tenth section was stained for Nissl substance with thionine. These sections were used to identify the location of dorsolateral prefrontal cortex area 46 by cytoarchitectonic criteria
(15,
16). From a series of sections determined to contain area 46, we chose four sections separated by 400 µm, beginning with a random starting point. Tissue sections were treated with 1% hydrogen peroxide for 15 minutes to eliminate endogenous peroxidase activity; they were then processed with the use of a rabbit anti-GAT-1 antibody (1:1000 dilution; Chemicon, Temecula, Calif.) and the Vectastain Elite ABC kit as previously described
(8). Sections from each triad of subjects were processed together through all steps and were mounted on coded slides. The specificity of the anti-GAT-1 antibody has been previously demonstrated
(8,
17).
Quantification of GAT-1-Immunoreactive Cartridges
All quantification was performed without knowledge of subject number or diagnosis. GAT-1-immunoreactive cartridge density was measured in the superficial layers (2–3a), middle layers (3b–4), and deep layer (layer 6) of area 46. These layers were selected for analysis because 1) they contain the highest densities of GAT-1-immunoreactive cartridges in normal subjects, and 2) the pyramidal cells in these layers have different patterns of afferent and efferent connections
(11). Laminar boundaries were determined by measuring the distance from the pial surface to the borders of each layer on Nissl sections and calculating the location of each border as a percentage of total cortical thickness. On average, the layer 1–2 border was located at 10%, the layer 3a–3b border at 35%, the layer 4–5 border at 60%, and the layer 5–6 border at 80% of the distance from the pial surface to the white matter. On each section, three separate counting regions (corresponding to the superficial, middle, and deep layers) were outlined with the Stereo Investigator Fractionator program (MicroBrightfield, Inc., Colchester, Vt.). A sampling grid (350 µm × 350 µm) was randomly placed over each counting region, and all cartridge-like profiles that fell within the sampling frames (225 µm × 225 µm) were counted at a final magnification of 400× by using rules for unbiased counting. The average Scheaffer coefficients of error (calculated by the Fractionator program) were 11.3%, 13.2%, and 13.0% for the normal, schizophrenic, and other psychiatric subjects, respectively. When corrected for sampling frames located along the exclusion boundaries for each of the counting regions, these values decreased by 15%–30%, resulting in coefficients of error of approximately 10% for each cortical layer.
The procedures described above were used for the 45 subjects (triads 16–30) that were new to this study. For triads 1–15, relative cartridge density was assessed by using a cortical traverse counting procedure as previously described
(8). Direct comparison has shown that the two procedures produce similar density estimates
(8). For triads 1–15, cartridge density values had previously been reported for the entire cortical thickness. For the present study, the subjects from triads 1–15 were reanalyzed by applying the laminar percentages described above to the cortical depth coordinates of the cartridges in order to obtain density measures of GAT-1-immunoreactive cartridges in layers 2–3a, 3b–4, and 6. Mean densities of GAT-1-immunoreactive cartridges did not differ between the subjects in triads 1–15 and triads 16–30 in layers 2–3a (mean=83.2/mm
2, SD=47.0, and mean=72.5/mm
2, SD=40.5, respectively), layers 3b–4 (mean=47.6/mm
2, SD=36.7, and mean=45.1/mm
2, SD=26.1), and layer 6 (mean=98.4/mm
2, SD=49.9, and mean=111.8/mm
2, SD=51.9) (F<1.55, df=1, 88, p>0.22).
Assessment of the Effect of Antipsychotic Medications on GAT-1-Immunoreactive Cartridges
To assess the influence of psychotropic medications on the density of GAT-1-immunoreactive cartridges, we studied four male cynomolgus (
Macaca fascicularis) monkeys treated chronically with haloperidol decanoate. Each drug-treated animal was matched to an untreated comparison animal on sex, age, and weight. The animals initially received daily doses of haloperidol in order to determine the amount required to produce 8-hour trough serum levels of 4–8 ng/ml—levels shown to be associated with a therapeutic response in humans
(18). As per standard clinical practice, that dose was multiplied by 15 to obtain the dose of haloperidol decanoate to be administered intramuscularly every 4 weeks. The mean dose of haloperidol decanoate was 16.0 mg/kg (SD=2.1), and the mean trough serum level obtained just before the next dose was 4.3 ng/ml (SD=1.1). Consistent with our attempt to mimic the clinical model of “neuroleptic threshold dosing”
(19), all haloperidol-treated animals developed mild extrapyramidal symptoms that were controlled with maintenance benztropine mesylate (1 mg b.i.d.). After 9–12 months of treatment, both animals in a matched pair were euthanized with an overdose of pentobarbital, and the brains were removed. All procedures were approved by the University of Pittsburgh’s Institutional Animal Care and Use Committee.
After a 45-minute postmortem interval, coronal tissue blocks from the left hemisphere were prepared and processed for GAT-1 immunocytochemistry in a manner identical to that described for the human subjects. The densities of GAT-1-immunoreactive cartridges in layers 2–3a and 3b–4 of area 46 were determined in a fashion identical to that described above with one exception. Because of the thinner cortical mantle in monkeys, the sampling frames (190 µm × 190 µm) were randomly placed at intervals of 225 µm in each counting region. The coefficient of error in each sampling zone of each animal was less than 10%.
Statistical Analysis
For the human studies of GAT-1-immunoreactive cartridge density, regression analyses revealed a significant association between cartridge density and age, but not sex, postmortem interval, or tissue storage time. Because the last three measures also did not differ between subject groups (
table 2), differences in cartridge density within each layer were analyzed with the use of a general linear model based on a complete randomized block design, with diagnostic group, triad (block), and age as independent variables
(20). Analyses were done in PROC GLM in SAS software, and F ratios were interpreted with type III sum of squares. Post hoc pairwise comparisons with the schizophrenic group as the target were made by means of Dunnett’s adjustment for multiple comparisons, as this test controls type I experimentwise error. To examine the magnitude and variability of these comparisons, Dunnett’s t tests were used with an alpha level of 0.05 to calculate confidence intervals. For the monkey studies, analysis of covariance, with body weight as a covariate, was used to assess group differences in cartridge density for each layer.
RESULTS
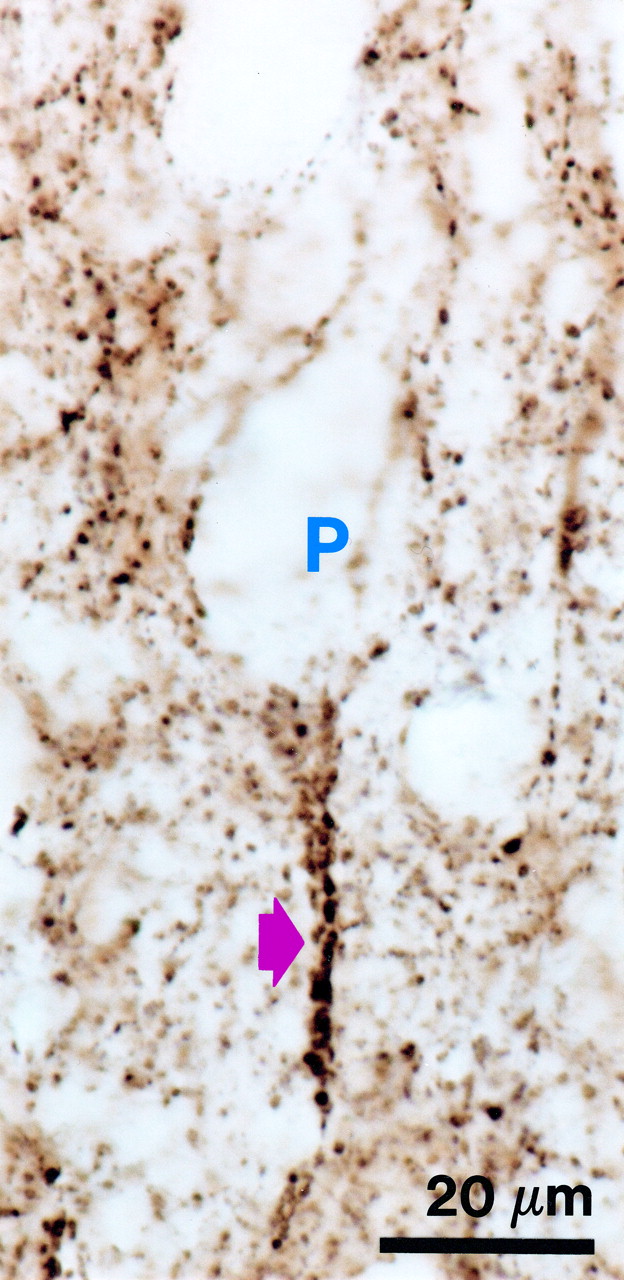
As previously reported
(8), GAT-1-immunoreactive cartridges were readily distinguished from other immunoreactive punctate structures by their characteristic appearance and location below the soma of unlabeled pyramidal cells (
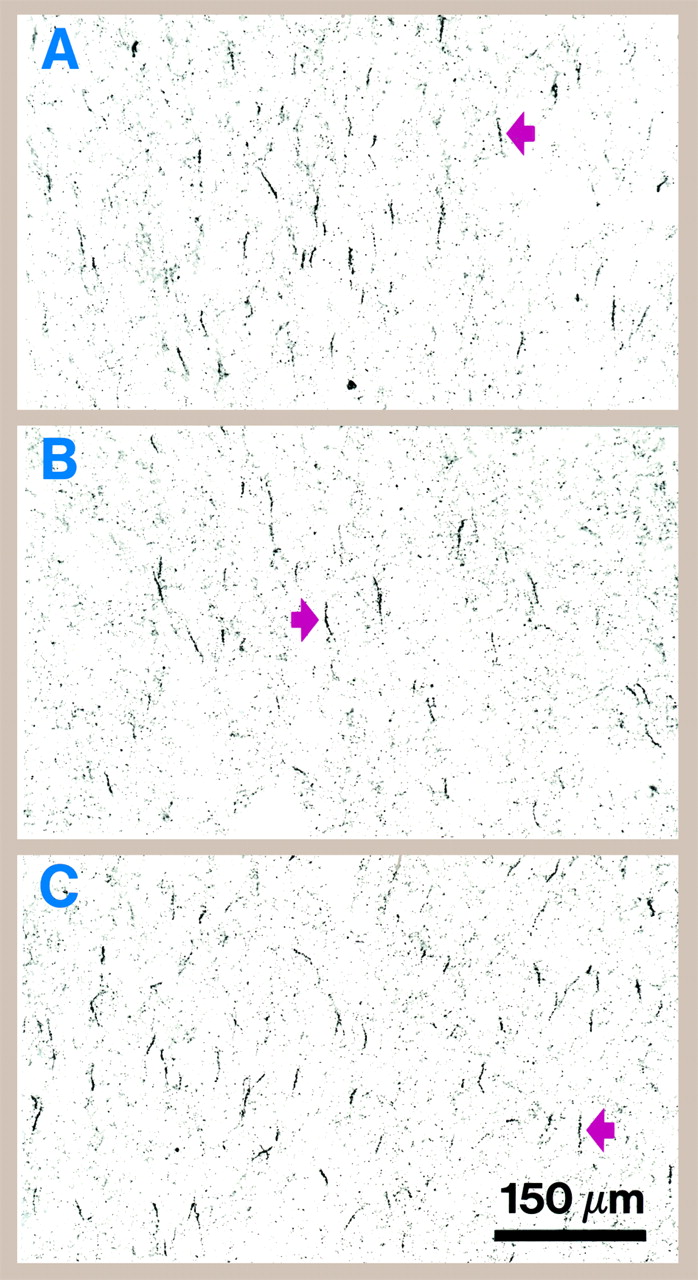
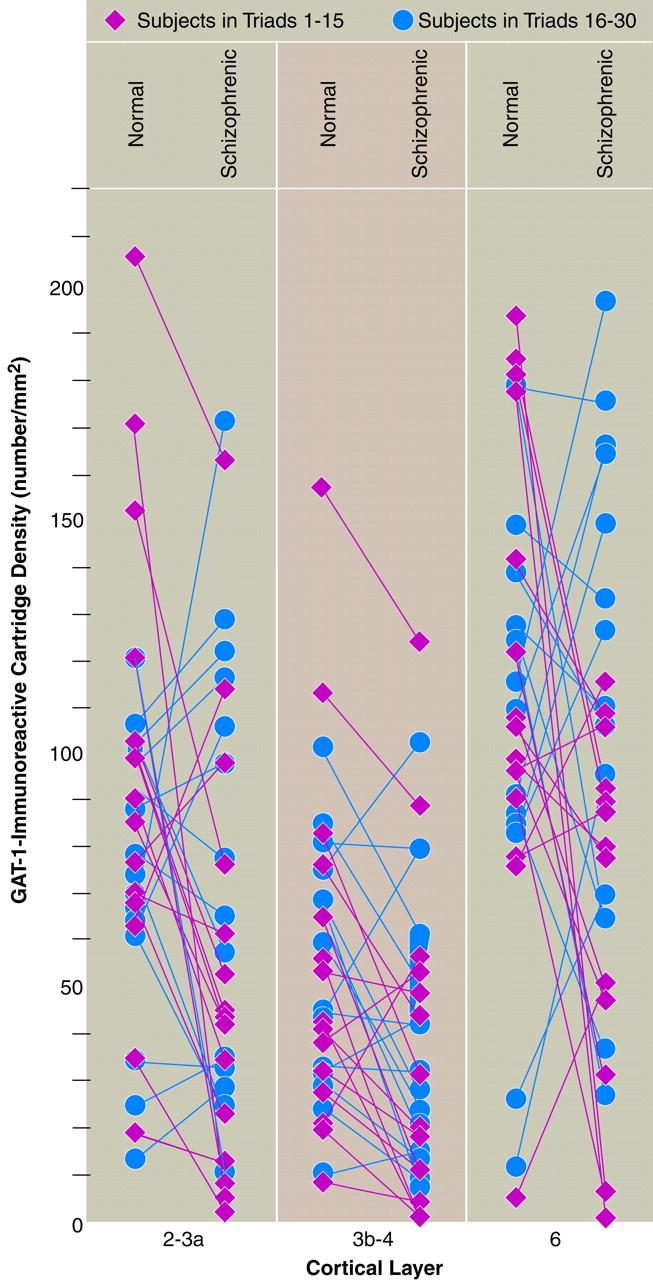
figure 1). In some triads of subjects, a lower density of labeled cartridges in the schizophrenic subject relative to the matched comparison subjects was evident by qualitative inspection (
figure 2).
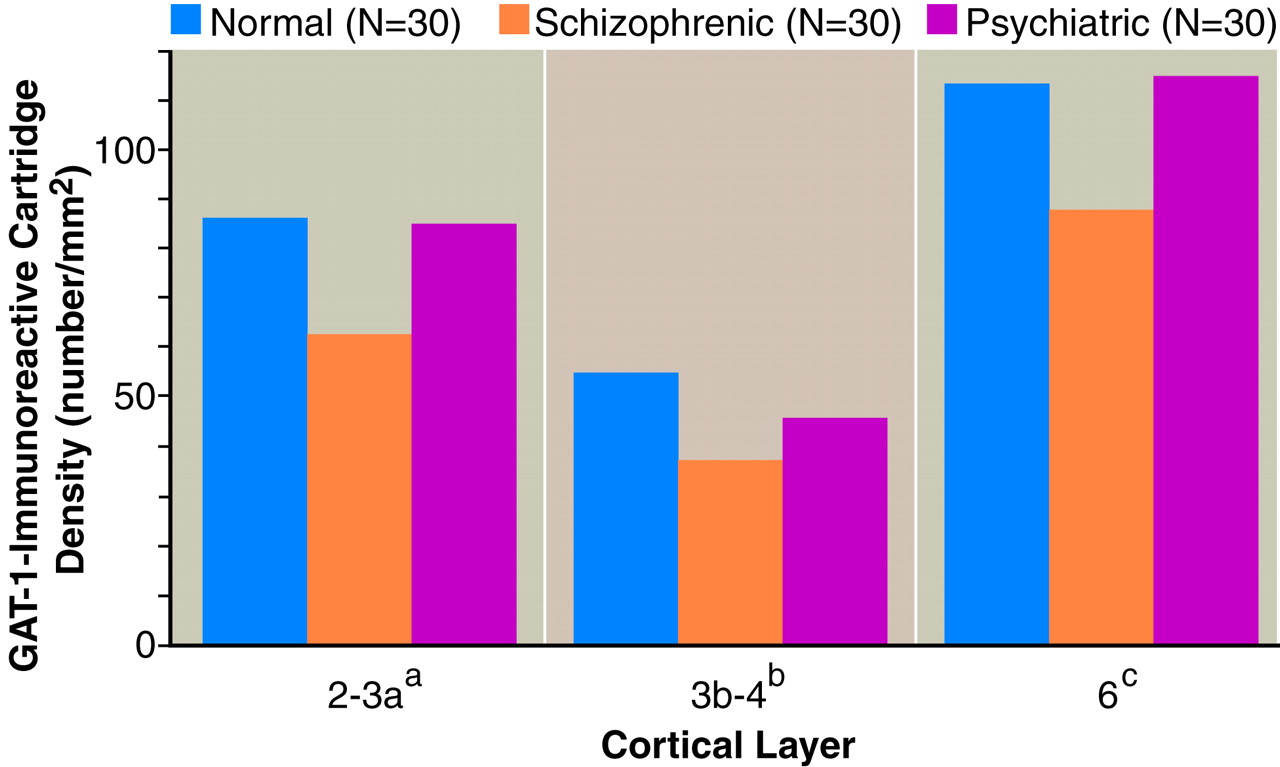
Comparison of GAT-1-immunoreactive axon cartridge density in area 46 across the three groups of human subjects (
figure 3) revealed a significant effect of diagnosis in the superficial and middle layers, whereas in the deep layers, the group differences did not quite achieve statistical significance. In contrast, the effect of age was not significant in any layer (F<2.70, df=1, 57, p>0.11). In the superficial layers, mean cartridge density in the schizophrenic subjects was significantly (p<0.05) decreased by 27.7% (95% confidence interval [CI]=2.4%–53.0%) and 26.4% (95% CI=1.3%–51.5%) in comparison with the matched normal and psychiatric comparison subjects, respectively. Similarly, mean cartridge density in the middle cortical layers was decreased in the schizophrenic subjects by 31.5% (95% CI=9.0%–54.0%) relative to the normal comparison subjects and by 19.4% (95% CI=–6.9%–45.7%) relative to the psychiatric comparison subjects, although the latter difference did not achieve statistical significance. Cartridge density did not significantly differ between the normal and psychiatric comparison subjects in any of the three layers.
Analysis of the data for the individual pairs of schizophrenic and normal subjects suggested that the decrease in cartridge density in the schizophrenic subjects might be more common in the middle cortical layers than in the superficial or deep layers. For example, cartridge density was decreased in the schizophrenic subjects in 80.0% of the pairs in layers 3b–4 (compared with chance expectations, χ
2=5.9, df=1, p=0.01) but in only 66.7% of the pairs in layers 2–3a (χ
2=1.7, df=1, p=0.19) and 63.3% of the pairs in layer 6 (χ
2=1.1, df=1, p=0.30) (
figure 4). In addition, the frequency and magnitude of the decrease in cartridge density in the middle layers was similar across the two groups of subjects (triads 1–15 and 16–30). For example, in layers 3b–4 of the former group, 13 of 15 schizophrenic subjects showed a decrease in GAT-1-immunoreactive cartridge density relative to their matched normal comparison subjects, and this decrease averaged 55.2% (SD=28.6%). Similarly, in the same layers of the latter group, cartridge density was decreased in comparison with the matched comparison subjects in 11 of 15 schizophrenic subjects, with a mean decrease of 44.0% (SD=28.7%) in these subjects (
figure 4).
There was a significant main effect of diagnosis on cortical thickness (F=3.40, df=2, 58, p=0.04). Post hoc comparisons revealed a significant (p<0.05) 5.9% decrease in the mean cortical thickness of the psychiatric comparison subjects (mean=2602 µm, SD=235) relative to that of their matched normal comparison subjects (mean=2764 µm, SD=267). In contrast, cortical thickness in the subjects with schizophrenia (mean=2697 µm, SD=315) was not significantly different from that of either comparison group.
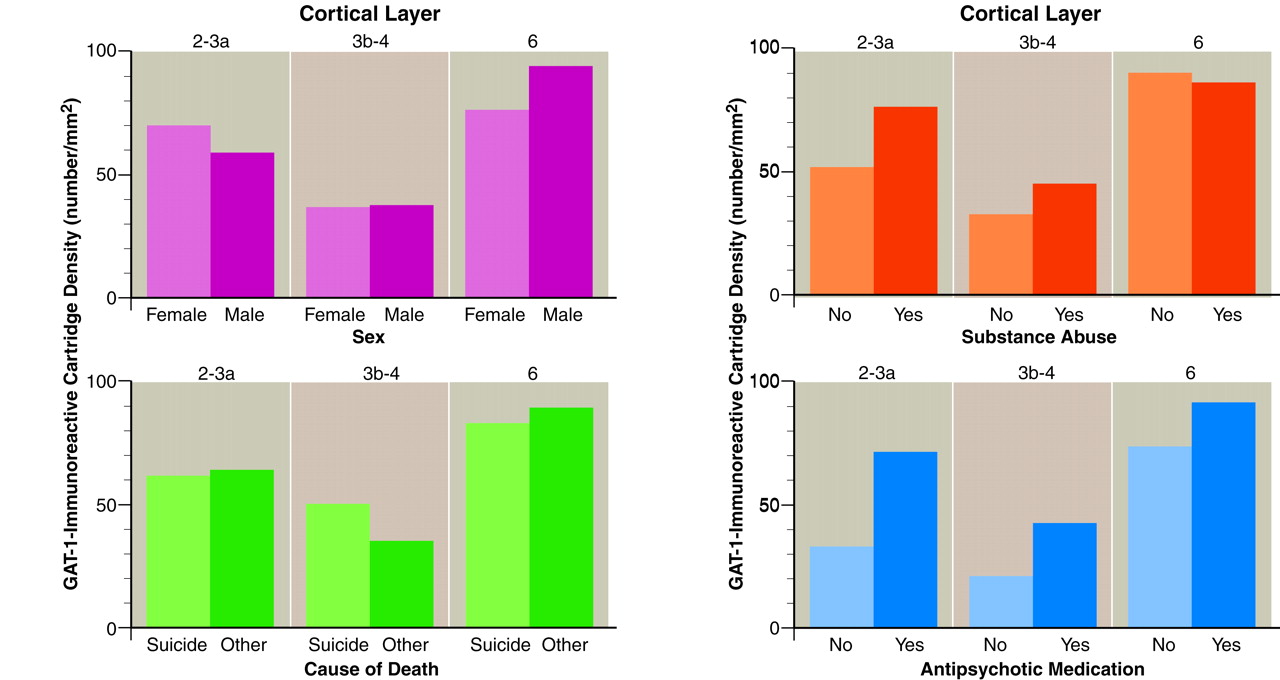
To explore the effects of age at onset and duration of illness on GAT-1-immunoreactive cartridge density, we used multiple regression analyses. The subject’s age at the time of death and either age at illness onset or duration of illness were included as independent variables, since age at the time of death was correlated with both of these variables. Neither age at onset nor duration of illness was significantly associated with GAT-1-immunoreactive cartridge density in any layer in either the schizophrenic or the other psychiatric subjects (all partial correlation p values >0.50). In addition, cartridge density in the schizophrenic subjects did not significantly differ as a function of sex, suicide, history of alcohol or substance abuse, or antipsychotic treatment status at the time of death in any of the cortical layers examined (
figure 5).
To evaluate whether the observed decrease in GAT-1-immunoreactive cartridges in schizophrenic subjects might be a consequence of treatment with psychotropic medications, a similar study was performed in the dorsolateral prefrontal cortex of monkeys chronically treated with haloperidol and benztropine. In contrast to the decreased density of GAT-1-immunoreactive cartridges observed in the schizophrenic subjects, mean cartridge density was increased by 21.6% in layers 2–3a (haloperidol-treated animals: mean=337/mm2, SD=76.0; control animals: mean=279/mm2, SD=33.5) and by 9.8% in layers 3b–4 (haloperidol-treated: mean=291/mm2, SD=53.8; control: mean=269/mm2, SD=72.1) of the haloperidol-treated animals in comparison with their matched control subjects, although these differences were not significant (F=1.59, df=1, 5, p=0.26, and F=6.31, df=1, 5, p=0.05, respectively).
DISCUSSION
The results of this study demonstrate that the density of chandelier neuron axon cartridges immunoreactive for GAT-1 is decreased in dorsolateral prefrontal cortex area 46 in a majority of subjects with schizophrenia. Comparison with subjects with other psychiatric disorders suggests that decreased GAT-1 cartridge density may be specific to the disease process of schizophrenia, or at least that it is not a common correlate of psychiatric illness, suicide, comorbid substance abuse or dependence, or exposure to antipsychotic medications. In addition, our results suggest that a decrease in GAT-1-immunoreactive cartridge density may be most pronounced and common in the middle cortical layers, raising the possibility that a subset of chandelier neurons and their target pyramidal neurons are preferentially affected in schizophrenia. The laminar location of this abnormality suggests that pyramidal neurons participating in corticocortical circuitry
(11) and thalamocortical circuitry
(21,
22) may be most affected. Interestingly, these circuits appear to be involved in working memory
(23), a fundamental cognitive function that is disturbed in schizophrenia
(1,
2).
Methodological Issues
We report relative densities as opposed to absolute numbers of GAT-1-immunoreactive cartridges in this study, since the latter measure was neither feasible nor necessary
(24). To obtain an estimate of the absolute number of cartridges in area 46, the volume of this cytoarchitectonic region for each subject must be accurately identified. However, because boundaries between cytoarchitectonic areas are not sharp and involve extensive transition zones in the human dorsolateral prefrontal cortex
(15,
16), the total number of cartridges in area 46 cannot be reliably determined. In addition, the thickness of tissue sections may shrink by 70% during processing for immunocytochemistry, interfering with the three-dimensional sampling required for estimation of absolute numbers. Furthermore, two potential limitations of the use of profile-counting procedures to determine relative density (differences in cartridge size or cortical volume across diagnostic groups) do not appear to have been confounding factors in the present study. We previously demonstrated that the length of GAT-1-immunoreactive cartridges did not differ across the subject groups in triads 1–15
(8). In addition, the subjects with schizophrenia in the present study showed no increase and perhaps a slight decrease in cortical thickness. This decrease in cortical thickness, consistent with reports of decreased dorsolateral prefrontal cortex volume in schizophrenia
(25–
27), suggests that the decrease in GAT-1-immunoreactive cartridges in schizophrenic subjects may actually be greater than that reported in the present study. Finally, the use of systematic random sampling techniques and assessments of sampling adequacy in this study ensured that the measures obtained were reliable estimates of relative cartridge density in each subject.
The three groups of subjects were also matched on variables critical to interpreting postmortem studies, such as postmortem interval and tissue storage time, neither of which was correlated with cartridge density. In addition, although the subjects with schizophrenia differed from the other psychiatric subjects on measures of age at illness onset and duration of illness, these variables did not correlate with GAT-1-immunoreactive cartridge density independent of age, suggesting that length of illness does not account for the differences in GAT-1-immunoreactive cartridge density between these two groups. Finally, decreased GAT-1-immunoreactive cartridge density was not associated with sex, suicide, or diagnosis of an alcohol or substance use disorder within the group of subjects with schizophrenia, suggesting that these variables did not affect our findings.
Influence of Antipsychotic Medications on GAT-1-Immunoreactive Cartridge Density
Understanding the significance of decreased cartridge density depends on whether this decrease is integral to the pathophysiology of schizophrenia or a result of treatment of the illness. We previously reported that the regional densities of GAT-1-immunoreactive cartridges did not differ between normal comparison subjects and nonschizophrenic psychotic subjects treated with antipsychotic medications
(8). These findings suggested that the observed decrease in cartridge density in schizophrenic subjects was not a consequence of medical treatment. Three observations from the present study confirm this interpretation. First, in the schizophrenic subjects who were free of antipsychotic medications at the time of death or had never received them, average GAT-1-immunoreactive cartridge density was less than that of their matched normal comparison subjects across all cortical layers. Second, cartridge density did not differ between schizophrenic subjects on or off antipsychotic medications at the time of death (
figure 5). Third, the density of labeled cartridges was not decreased in the dorsolateral prefrontal cortex of monkeys treated for 9–12 months with haloperidol and benztropine.
Biological Basis for Decreased GAT-1-Immunoreactive Axon Cartridge Density
Although the decreased density of GAT-1-labeled axon cartridges might be due to a decrease in the number of chandelier neurons, the majority of studies have failed to detect a decrease in the number or density of prefrontal neurons in schizophrenia
(5,
15,
28,
29). Moreover, neither the density nor the laminar distribution of neurons containing parvalbumin, a protein present in chandelier neurons
(30,
31), was altered in the dorsolateral prefrontal cortex of 15 schizophrenic subjects (triads 1–15) examined in the present study
(32).
Since the typical chandelier neuron furnishes 200–300 cartridges
(9), it is possible that the axon arbor of these neurons is altered in schizophrenia such that each neuron gives rise to fewer cartridges. Such a reduction could occur in response to a decrease in the density of pyramidal neurons, the targets of chandelier neuron axon cartridges. However, consistent with other reports
(29), the density of Nissl-stained pyramidal neurons in deep layer 3 was not decreased in the same 30 schizophrenic subjects examined in the present report
(33). Furthermore, the size of parvalbumin-positive neurons, which includes the chandelier class, does not appear to be decreased in schizophrenia
(32). Since a neuron’s somal size tends to be correlated with the total length of its axon
(34,
35), these findings are not consistent with the notion that the axon arbor of chandelier neurons is reduced in schizophrenia.
Finally, a decrease in GAT-1 mRNA expression in chandelier cells could result in low (or absent) levels of the protein in axon cartridges so that they are undetectable by immunocytochemical techniques. For example, during postnatal development of primate dorsolateral prefrontal cortex, other protein markers for axon cartridges decline substantially
(36), without an apparent loss of terminals
(37) or a change in the number of inhibitory synapses
(38). In addition, expression of the mRNA for GAD
67, the synthesizing enzyme for GABA, is reduced in schizophrenia
(5), and this change appears to be restricted to a subpopulation of GABA neurons, especially those located in the middle cortical layers
(6). Together, these findings support the interpretation that the decrease in density of GAT-1-immunoreactive axon cartridges in schizophrenia represents a diminished amount of this protein in chandelier neurons. However, further testing of this hypothesis is necessary.
Pathophysiological Significance of Decreased GAT-1-Immunoreactive Axon Cartridges
If the findings of the present study reflect a decrease in GAT-1 protein in axon cartridges, then this abnormality may represent a defect in chandelier cells such that they either underexpress GAT-1 mRNA or do not produce the GAT-1 protein or direct it to the axon terminal in a normal manner. Similar to other neurotransmitter membrane transporters, GAT-1 plays a central role in regulating the magnitude of effect and duration of action of GABA at postsynaptic sites
(39,
40). Although less is known about the precise function of GAT-1, the dopamine membrane transporter appears to be the critical determinant of the availability of dopamine in the extracellular space
(41). If GAT-1 functions similarly, then decreased GAT-1 protein in axon cartridges would be associated with increased GABA concentrations and enhanced inhibition at the axon initial segment of pyramidal cells. Indeed, in the dopamine membrane transporter knockout mouse, the absence of dopamine membrane transporter is associated with excess dopamine activity and a compensatory down-regulation of tyrosine hydroxylase, the rate-limiting enzyme in the synthesis of dopamine
(41). As noted above, the expression of GAD
67 mRNA is decreased in the dorsolateral prefrontal cortex of schizophrenic subjects
(5), and this decrease appears to be selective for a subpopulation of GABA neurons
(6). By analogy to the dopamine system, these findings suggest that GAD
67 mRNA expression, and consequently GABA synthesis, may be down-regulated in chandelier neurons in order to compensate for excessive GABA activity at the axon initial segment of pyramidal cells. The notion that decreased GAT-1 represents a “primary” defect of chandelier neurons may be supported by the observation that both schizophrenic subjects on medication at the time of death and haloperidol-treated monkeys showed a trend toward an increase in the density of GAT-1-immunoreactive axon cartridges in the dorsolateral prefrontal cortex (
figure 5). These observations suggest that some of the beneficial effects of antipsychotic medications may occur by reversing a GAT-1 deficiency.
Alternatively, decreased density of GAT-1-immunoreactive axon cartridges in schizophrenia may reflect a “secondary” response to altered afferent input to the dorsolateral prefrontal cortex. For example, the number of neurons in the mediodorsal thalamic nucleus has been reported to be decreased in schizophrenia
(42–
44). Projections from this nucleus, the principal source of excitatory thalamic afferents to the dorsolateral prefrontal cortex, terminate primarily on the dendritic spines of pyramidal neurons in layers deep 3 and 4
(21,
22). In addition, the density of dendritic spines, markers of excitatory inputs, is decreased on the basilar dendrites of dorsolateral prefrontal cortex pyramidal neurons
(45,
46), especially those located in layers deep 3 and 4
(46). Together, these findings support the hypothesis that the excitatory thalamic drive to dorsolateral prefrontal cortex pyramidal cells is diminished in schizophrenia and raise the possibility that the reported decrease in dorsolateral prefrontal cortex GAD
67 mRNA expression represents a compensatory down-regulation. For example, in primary visual cortex, decreased activity in thalamic inputs produced a decrease in GAD
67 expression that was most marked in the layers receiving these inputs
(47). Although we are not aware of any data directly addressing this issue, if GAT-1 expression is co-regulated with GAD
67, our findings of a more prominent decrease in the density of GAT-1-immunoreactive cartridges, and in the density of neurons that express GAD
67 mRNA
(6), in the middle layers of the dorsolateral prefrontal cortex in subjects with schizophrenia may represent a compensation for diminished input from the mediodorsal thalamic nucleus. Differentiating between these possible explanations for decreased GAT-1-immunoreactive axon cartridges in schizophrenia may provide further insight into the pathophysiology of the debilitating cognitive symptoms of this disorder.