Cocaine abuse can cause strokes
(1,
2). The relative likelihood of developing a stroke in cocaine users may be as much as 14 times greater than that in age-matched non-cocaine-using comparison subjects recruited from the same patient pool
(3). Between 25% and 60% of cocaine-induced strokes can be attributed to cerebral ischemia
(4–
6). About 80% of the infarcts occur in the regional distribution of the middle cerebral artery
(7), typically in young adults without preexisting vascular malformations
(6). Nevertheless, increased polysubstance abuse among cocaine addicts has complicated attribution and accurate characterization between amount and duration of cocaine use and its relationship with global and regional hypoperfusion abnormalities
(8). Perfusion deficits also can occur in the absence of clinically detectable symptoms in the central nervous system
(9,
10) and may persist in abstinent cocaine addicts for 6 months
(11) or more
(12). Mechanistic processes that underlie cocaine’s site-specific effects on the cerebral vasculature are, therefore, imperfectly understood. Advanced neuroimaging techniques offer a vehicle for elucidating these mechanisms and ascertaining the effectiveness of putative therapeutic agents for treating cerebral ischemia.
Preclinical research suggests that the etiology of cocaine-induced brain ischemia is multifactorial and involves the stimulation of vasospasm
(13), platelet aggregation
(14), and pathological changes in the cerebral vasculature that can impair cellular oxygenation
(14). Within all of these processes, calcium-channel-mediated interactions, particularly with dopamine, play an important role. Dihydropyridine-class calcium channel antagonists, by counteracting cocaine-induced vasospasm, have potential as putative therapeutic agents
(15).
This review article is subdivided into three major parts. First, we critically appraise the neurobiology of cocaine-induced cerebral ischemia. Second, we summarize current knowledge on the sequelae of cocaine’s neuroischemic effects. Third, we assess the role of various putative therapeutic agents for treating cocaine-induced cerebral ischemia, with a particular focus on dihydropyridine-class calcium channel antagonists.
Neurobiology of Cerebral Ischemia
Cerebral ischemia is associated with a variety of cocaine’s neurovascular effects. We will review the relative importance of various mechanistic processes attributable to cerebral ischemia.
Vasospasm
Cocaine, the alkaloid benzoylmethylecgonine, by being highly lipid soluble, traverses the blood-brain barrierrapidly. As in the periphery, cocaine blocks monoamine reuptake, particularly those related to dopamine
(7). Cocaine-induced changes in dopamine level are most prominent in mesocorticolimbic neurons
(16,
17). Augmentation of mesocorticolimbic dopamine function may be associated with cerebral ischemia by means of two mechanisms. First, dopamine may control local blood flow by inducing vasospasm of smooth muscles lining the cerebral vessels
(18–
21), particularly those of the middle cerebral artery
(19). Second, cocaine-induced reductions in cerebral metabolism may lead to feedback down-regulation of blood flow
(22). Rapid reperfusion of these previously ischemic areas may, conversely, result in hemorrhage
(23).
More recently, the role of dopamine in the regulation of cortical blood flow has been better elucidated. First, my colleagues and I
(24) established that cocaine-induced cerebral vasospasm was relatively specific to dopamine-rich brain areas and hypothesized that dopamine pathways play a central role in controlling cerebral blood flow (CBF). Later, Krimer and colleagues
(25) provided a mechanistic basis for these findings by showing that dopamine-containing neurons were located adjacent to blood vessels in the frontal lobe and prefrontal cortex. These vessels constricted in response to the perivascular application of dopamine with the maximal calculated reduction of up to 58% of pretreatment flow. Hypothetically, in the living brain, pericytes containing contractile elements located proximal to dopamine nerve endings, which are also responsive to vasoactive substances in the endothelium, may be mechanistically responsible for this process
(26,
27). Essential to this process appears to be an initial transmembrane loss of magnesium followed by a rapid rise in intracellular calcium concentration. Severity of the perfusion deficit is, therefore, typically correlated with the relative strength of these neurochemical changes
(28). Nevertheless, cocaine’s vasospastic effects can persist beyond its half-life because its major metabolites (benzoylecgonine and norcocaine) also are potent vasoconstrictors
(29), and bradykinin-mediated endothelium-dependent relaxation is impaired in chronic cocaine abusers
(30,
31).
Although intracortical vessels are preferentially innervated by dopamine, those located extraparenchymally receive their neuronal supply from norepinephrine-containing cells in the superior cervical ganglia
(25). Thus, multifactorial mechanisms may be associated with the development and extension of cerebral vasoconstriction. Deep cortical brain regions may be more susceptible to dopamine’s vasospastic effects. In contrast, reductions in CBF subsequent to constriction of large cranial arteries may be more dependent on norepinephrine facilitation
(32,
33). The vasospastic effects of serotonin (5-HT), levels of which are also increased by cocaine, also may be greater at large and medium-sized cranial arteries. Cocaine’s vasospastic effects can, therefore, be associated with generalized global reductions in CBF
(34).
Thrombosis
Cocaine-induced cerebral ischemia also can be caused by thrombus formation in the vasculature
(29). Chronic cocaine use increases platelet levels, enhances adenosine diphosphate platelet activation, and augments sporadic release of platelet-bound α granules. Platelet activation and aggregation, precursors of thrombus formation, may be mediated by cocaine-induced increases in monoamine levels, particularly of 5-HT. Release of α granules containing growth factors and platelet-specific agglutinating proteins, β thromboglobulin and platelet factor 4, together with enhanced elastase-mediated atherosclerotic damage to the cerebral vasculature, all contribute to cocaine’s thrombogenic potential
(35,
36). Thrombus formation also may be facilitated by cocaine-induced cardiomyopathy
(14). In vitro studies also have demonstrated cocaine’s ability to facilitate the platelets’ response to arachidonic acid, thereby stimulating thromboxane production and, consequently, their aggregation
(37–
39). Protein C antithrombin III depletion and enhanced prostaglandin release are additional contributors to cocaine’s procoagulant effects
(40–
42). In sum, a gallimaufry of cocaine’s platelet aggregating and cardiomyopathic effects contribute to thrombus formation.
Vasculitis
Apart from the atherosclerotic changes attributable to chronic cocaine use that contribute to thrombus formation, there also is evidence of other pathological structural alterations in cerebral vasculature. For example, Martinez and co-workers
(14) found that long-term cocaine use, due to fluctuations between vasospasm and reperfusion, may result in vessel damage. This vasculitis, not attributable to arteriosclerosis or infection, has also been described by others
(2,
42). Chronic cocaine-induced vasculitis exacerbates nonlaminar blood flow and sludging in the vessels. This decreases cellular oxygenation, thereby increasing platelet aggregation and consequent thrombus formation.
Consequences of Cerebral Ischemia
Cerebral ischemia can result in a variety of quantifiable structural and functional deficits. These deficits contribute to the pathogenesis of cocaine dependence and can impair an addict’s recovery.
Although the focus of this review relates to the quantification of dynamic changes in CBF, it is noteworthy that structural defects also may be a long-term consequence of this abnormality. Ischemic cerebral damage can result in cellular death identifiable by magnetic resonance imaging (MRI). Cocaine-induced cerebral damage appears to have similar hallmarks to other diseases that result in global damage. For instance, as brain cells die, there are generalized atrophic changes with concomitant ventricular enlargement and sulcal widening
(42,
43). On a cellular level, ischemia-induced neuronal degeneration is associated with the extracellular leakage of glutamate. This is due to inactivation of the adenosine diphosphate-dependent glutamate reuptake pump, which results in the accumulation of intracellular sodium and consequent neuronal engorgement
(44). Glutamate’s neurotoxic effects are potentiated by the long-term influx of intracellular calcium. Excessive intracellular calcium concentration produces direct necrotic cellular changes and promotes the activity of calcium-sensitive proteolytic enzymes. Consequently, apoptotic processes may be triggered prematurely
(45). In the aggregate, cerebral ischemia can provoke or potentiate a sequence of events resulting in structural damage due to direct or programmed neuronal cell death.
Functional deficits represented as decreases in cerebral perfusion can exist in the absence of structural damage. These perfusion deficits have mainly been studied using two types of neuroimaging: single photon emission computed tomography (SPECT) and positron emission tomography (PET); however, future studies may make use of developing techniques in functional MRI.
The relationship between chronic cocaine abuse and reported cognitive deficits, hypothetically due to perfusion abnormalities in the striatum
(46), remains controversial. Methodological problems, such as small group size, use of published cognitive test norms instead of appropriately matched comparison subjects, differences in population characteristics (e.g., social class, treatment-seeking versus incarcerated status, presence or absence of concomitant drug abuse or dependence, varying lengths of abstinence, and level of care in its verification), have resulted in conflicting findings
(47,
48). Generally, controlled studies of cocaine abusers with several months of abstinence support an impairment of response in complex and simple psychomotor tasks
(47,
49) and to auditory tones or discrimination tasks
(50–
52) but less consistently demonstrate decrements in memory and attention
(47,
48). Future studies of the relationship between cocaine abuse and cognitive dysfunction need to address two current limitations in our knowledge. First, we know of no long-term longitudinal studies charting the relationship between the progression of disease, as identified by neuroimaging studies, and a comprehensive set of standardized cognitive tests. Second, it is important for us in more fully characterizing the relationship between cognitive function and functional brain abnormality to determine how these parameters are affected by whether or not the individual is actively taking cocaine, has recently withdrawn from it, or has entered into a period of prolonged abstinence. Development of accurate methods for characterizing functional changes in CBF, therefore, remains at the cornerstone of quantifying cerebral ischemia and its consequent neuropsychological sequelae.
Neuroimaging Findings
Neuroimaging studies have provided a vehicle for understanding cocaine’s ischemic effects on the brain. Early studies vary in the reported distribution of regional cerebral blood flow (rCBF) abnormalities found in cocaine users
(10,
53,
54); however, these rCBF deficits can be severe
(46,
55,
56). Although most abused substances (including cocaine) reduce both global and regional cerebral glucose metabolism during acute use, rCBF can be either increased or decreased, perhaps related to the vasoactive characteristics of particular compounds
(57). The distribution of both rCBF and abnormalities in regional cerebral glucose metabolism also depends on whether the individual was currently using the drug, was exposed to cues, had recently withdrawn from cocaine, or was in long-term detoxification
(53,
58,
59).
A typical report is that of Holman et al.
(60), who studied cocaine abusers with
99mTc-labeled exametazime ([
99mTc]hexamethylpropyleneamine oxime) and high-resolution SPECT. Sixteen of 18 cocaine-dependent users had abnormal perfusion characterized primarily as small focal defects involving the infraparietal, temporal and anterofrontal cortex, and basal ganglia (the results of psychometric testing were abnormal in all 18). There was no relationship between the severity of SPECT abnormalities and the mode of cocaine administration or the frequency or length of cocaine use. It is important, however, that these results of global and regional hypoperfusion were consistent with the findings of others
(61,
62). Later, Holman and colleagues
(58) reported that such findings can be confounded by similar vascular abnormalities observed in HIV-positive and AIDS patients because of the frequent comorbidity of HIV infection and drug abuse.
Pearlson and co-workers
(63) advanced these findings in a well-controlled study of acute cocaine dosing. They suggested that cocaine’s ischemic effects were most prominent in dopamine-rich brain regions such as the caudate, inferior cingulate, and frontal areas; however, these site-specific effects could not be established by this experiment because a quantitative approach to SPECT was not employed and the cocaine dose was not manipulated. Pearlson and co-workers
(63) also suggested that cocaine-induced reductions in CBF were associated with increased positive subjective mood and abuse liability. These findings differ from those of London and co-workers
(54), who, using PET and [
18F]fluorodeoxyglucose, did not find an association between regional cerebral glucose metabolism and subjective mood. An advantage of the study of Pearlson and co-workers
(63) was that imaging and the peak of cocaine’s behavioral effects were synchronized carefully, thereby increasing the predictive power of the findings. While a detailed description of the relationship between cocaine-induced subjective mood and other (i.e., non-flow-related) aspects of brain function is outside the scope of this review, further studies are clearly needed. Notably, however, this field of inquiry has been advanced by the demonstration that dopamine transporter function may be closely correlated with the expression of cocaine’s subjective effects
(64).
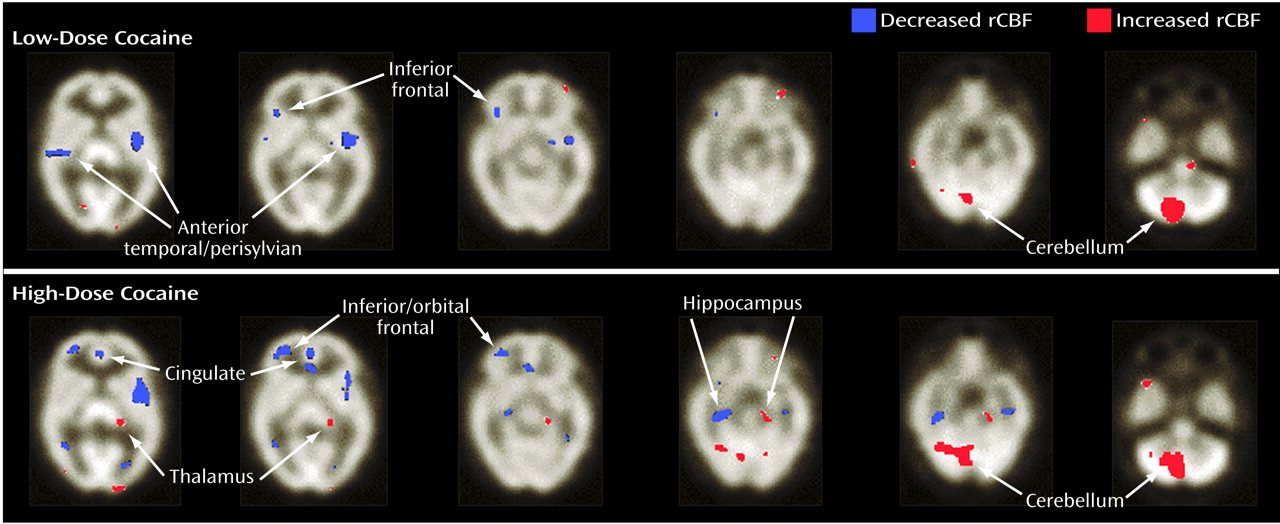
More recently, we studied the effects of cocaine on global and rCBF using a novel approach to quantified SPECT
(24). In that study, nine cocaine-dependent men and women (six men) with no evidence of structural brain abnormality (verified by an MRI at screening) were intravenously administered low and high doses of cocaine (0.33 mg/kg and 0.65 mg/kg, respectively) or placebo in a double-blind crossover fashion. We established that cocaine administration, compared with placebo, was associated with significant reductions in global and rCBF, which was relatively specific for dopamine-rich brain areas. Also, these CBF reductions were mostly dose dependent (i.e., the high dose led to greater involvement of cortical sites than the low dose), and the areas with greatest deficits were the prefrontal, frontal, temporal, and subcortical gray matter (
Figure 1). This study elucidated the site-specific effects of cocaine related to CBF changes and established the utility of this quantified approach to SPECT.
In sum, cocaine abuse, either acute or long-term, is associated with hypoperfusion-related CBF abnormalities. Despite some variance in the literature concerning the anatomical regions most affected, the current evidence suggests that dopamine-rich brain areas are particularly vulnerable. The correlation between regional blood flow reductions in dopamine-rich areas and cocaine abuse was intriguing and, as was later demonstrated, provided strong evidence for a role of cortical dopamine neurons in the regulation of the arteriolar microcirculation. Thus, medications that antagonize these dopamine-mediated effects on CBF offer promise as potential treatments for cocaine-related ischemic stroke.
Pharmacological Approaches
Until recently, despite the growing epidemic of cocaine-related ischemic stroke, the development of medications specifically targeted at this indication remained in its infancy. The next section describes the potential role of some promising medications aimed at reducing cocaine’s vasospastic, thrombogenic, or neurotoxic effects.
Antivasospastics
Animal studies support the use of the dihydropyridine-class calcium channel antagonist isradipine for the reversal of cerebral ischemia and the consequent neuronal cell damage
(65–
67). In a landmark study, Sauter and colleagues
(68) examined the relative effectiveness of various dihydropyridine-class calcium channel antagonists (i.e., darodipine, nimodipine, nitrendipine, and isradipine) in reducing the ischemic damage produced by occlusion of the middle cerebral artery in the rat. CBF was estimated by using the [
14C]iodoantipyrine method for blood flow quantification. In that study, isradipine (2.5 mg/kg given subcutaneously) had the greatest anti-ischemic effect. The dose-related vasodilator response to isradipine was wider than for the other dihydropyridine-class calcium channel antagonists and was correlated with a reduction in infarct size. These antivasospastic effects of isradipine were attributed to the blockade of dopamine release from cortical neurons involved in the control of the cerebral vasculature. In another example, 60 minutes of cerebral ischemia was induced in spontaneously hypertensive male rats by carotid artery occlusion. Hydrogen clearance was used to measure CBF, and in vivo microdialysis was used to quantify extracellular increases in dopamine and glutamate levels. Although the vehicle-treated group experienced 180- and 24-fold increases in extracellular dopamine and glutamate levels, respectively, isradipine significantly reduced the dopamine increase but had no effect on glutamate levels
(69). Ooboshi and colleagues
(70) confirmed these results. They showed that pretreatment with isradipine (200 μg/ml) did not alter ischemia-induced dopamine release in the striatum but produced a 37% decrease in dopamine levels in the forebrain. Additionally, isradipine significantly reduced forebrain ischemia (i.e., improved blood flow) but had no effect on striatal blood flow. Together, these results suggest that isradipine is a potent cerebral vasodilator with selectivity for dopamine-innervated brain regions.
Most recently, we extended these results to humans using a quantified SPECT approach by demonstrating that isradipine (10 mg given orally) administered 60 min before intravenous cocaine (0.33 mg/kg) prevented both global and regional ischemia in dopamine-rich brain areas
(15). This was the first clear demonstration that any medication, including isradipine, could prevent cocaine’s vaso-spastic effects on CBF. One caveat to this study was that there was no condition using isradipine alone. Thus, the relative strength of isradipine’s effectiveness at antagonizing cocaine-induced cerebral ischemia is unknown.
In sum, isradipine appears to be a promising therapeutic medication for the prevention of cocaine-related cerebral ischemia. It is important, however, that we await the results of larger-scale confirmatory neuroimaging studies and the results of clinical testing with the longitudinal evaluation of cerebral perfusion.
Antithrombogenic Agents
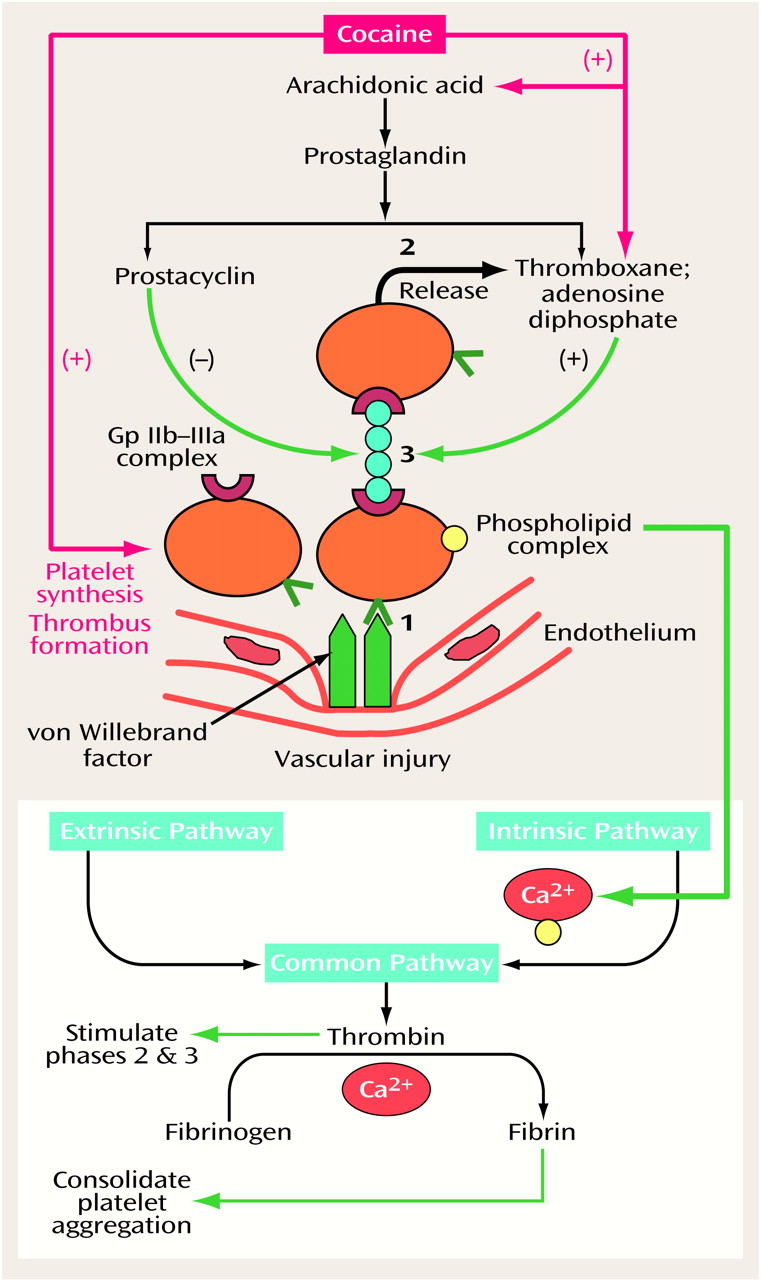
Recently, there has been interest in the use of aspirin to reduce cocaine’s thrombogenic effects. This is predicated on the finding that prostaglandin synthesis, which is enhanced by cocaine (
Figure 2), can be blocked by aspirin. Aspirin blocks this process by irreversibly inhibiting the action of cyclo-oxygenase in platelets released from bone marrow. These platelets, derived from the bone marrow, constitute about 15% of the body’s total daily circulation
(71,
72). Preliminary results indicate that aspirin (325 mg/day for 4 weeks) may improve CBF in chronic cocaine users; however, the results of a more definitive analysis are awaited
(73). Aspirin treatment may, however, increase the risk of spontaneous or uncontrolled bleeding from wounds. Thus, implementation of long-term aspirin treatment for cocaine addicts is impractical, because of their propensity for exposure to high-risk situations. Also, acute aspirin administration is unlikely to result in reversal of cerebral ischemia or substantial CBF improvement in individuals with an evolving ischemic episode of recent onset.
There may be potential for using the potassium-sparing diuretic amiloride for the prevention of ischemia caused by blockade of platelet α granule release in cocaine addicts
(73,
74). Again, the results of more definitive studies are awaited. Although the 5-HT
2 receptor antagonist ritanserin has demonstrated antiplatelet aggregation properties, its use for this indication in cocaine addicts remains unexplored
(75,
76).
Antineurotoxic Agents
One approach that has been suggested to reduce cocaine’s neurotoxic effects is the use of glutamate antagonists, such as phencyclidine and MK-801. Although these medications should, potentially, reduce the effects of excitatory amino acids released during neuronal degeneration at N-methyl-d-aspartate receptors, their other psychopharmacological effects limit their extensive use in humans. For instance, not only are these medications hallucinogenic, but they can also reduce the seizure threshold. Medications with antiglutamate properties and a more benign side effect profile, such as lamotrigine, or those that facilitate γ-aminobutyric acid neurotransmission, such as gabapentin, may have greater ability to prevent or treat cocaine-induced cerebral ischemia.
Evidence for the use of kappa opiate agonists for preventing ischemia because they modulate glutamate expression is conflicting (
77; cf. reference
78). Although decreases in cocaine-induced perfusion abnormalities have been reported with the kappa opiate antagonist buprenorphine, that study’s results were limited by the lack of a placebo control group
(59). Furthermore, research is needed to elucidate the role of kappa opiate antagonists in treating cocaine-induced cerebral ischemia.
Conclusions
Cocaine abuse significantly increases the risk of ischemic stroke. Current evidence shows that the genesis of cocaine-related ischemia is multifactorial. Principally, cocaine’s vasospastic effects at large cranial arteries or within the cortical microvasculature are mediated by increased levels of extracellular monoamines, particularly dopamine. Indeed, dopamine-rich brain areas appear to be relatively specific targets for cocaine-induced cerebral ischemia. The development of various neuroimaging tools, particularly PET and SPECT, has greatly advanced our knowledge about cocaine’s global and site-specific effects on CBF. Other mechanistic processes involved with the development of cerebral ischemia include thrombogenesis and vasculitis. Cocaine’s thrombogenic potential is associated with the enhancement of cellular processes that augment platelet aggregation and the enhancement of nonlaminar vascular flow after the development of cardiomyopathy.
Cocaine-induced cerebral ischemia can result in marked hypoperfusion abnormalities. These abnormalities may themselves be associated with deficits in the manipulation of complex and simple psychomotor tasks, but the evidence supporting memory and attentional deficits is less established. Little is known about the pathological development of these hypoperfusion abnormalities, but the prevailing evidence suggests that they persist even after a prolonged period of cocaine abstinence. With repeated cerebral ischemic episodes and subsequent reperfusion, vascular walls are weakened, and the likelihood of hemorrhagic episodes increases. Severe cerebral ischemia can result in direct neuronal death and degradation with the liberation of excitatory amino acids, particularly glutamate, which itself potentiates this process. Antithrombotic agents or medications that can ameliorate cocaine’s neurotoxic effects may have use in preventing or treating cocaine-induced cerebral ischemia. Promising recent data provide strong support for the use of dihydropyridine-class calcium channel antagonists in preventing cocaine-induced vasospasm, the principal cause of cerebral ischemia. Thus, the use of dihydropyridine-class calcium channel antagonists may herald a new vista in the prevention of cocaine-induced cerebral ischemia.