Major depressive disorder is a debilitating and, in some cases, potentially life-threatening psychiatric disorder. Major depression affects an estimated 350 million people worldwide and is a leading cause of years lost due to disability (
1). However, despite the high prevalence and immense economic burden associated with major depression, relatively little is known about the pathophysiology underlying the disorder. Although the monoaminergic neurotransmitter systems have been the primary focus in the majority of investigations exploring the associated pathophysiology for the past five decades, evidence is rapidly accumulating to suggest that the amino acid neurotransmitter systems and impaired mitochondrial function contribute to pathophysiological mechanisms of mood and other stress-related disorders (
2,
3).
Clinical studies using magnetic resonance spectroscopy (MRS) have repeatedly shown abnormal amino acid neurotransmitter (glutamate, glutamine, and GABA) content in the brains of patients diagnosed with mood disorders. A meta-analysis of proton (
1H) MRS studies found evidence of both diffuse and region-specific changes in glutamine and glutamate content (
4), and the recent discovery of significant antidepressant effects associated with drugs targeting the glutamatergic system has led to speculation on the contributions of the system to the pathophysiology of mood disorders (
5). In addition to evidence linking glutamate to mood disorders, there is a long history of studies suggesting that abnormalities in the GABA-ergic system contribute to the pathophysiology of mood disorders (
6). Early reports suggested that GABA levels were reduced in the blood and cerebrospinal fluid of patients with depression (
7). Imaging studies have provided fairly consistent reports suggesting that GABA content is reduced in the brains of patients with depression, especially in the occipital cortex (
8,
9), although this appears to occur primarily in certain subtypes of the disorder. Additional studies have suggested that altered function of the amino acid neurotransmitter systems contributes to changes in cortical excitability and inhibition as well as several other pathophysiological processes related to mood disorders (
10,
11).
One potential pathophysiological mechanism that could be related to the findings of altered amino acid neurotransmitter system content and function in individuals with mood disorders is impairment in astrocyte function. Astrocytes provide a primary means of glutamate and GABA clearance through the excitatory amino acid transporters 1 and 2 and GABA transporters 1–3, respectively (
12,
13). Astrocytes also play a critical role in the metabolism of both glutamate and GABA. Once glutamate is brought into an astrocyte, it is rapidly converted into glutamine by glutamine synthetase. The glutamine is then transported back to glutamatergic neurons to replenish glutamate stores by the mitochondrial enzyme glutaminase in a process referred to as the glutamate/glutamine cycle (
14). Glutamine is also transported to GABA-ergic cells, where it is first converted into glutamate by glutaminase and then to GABA by glutamic acid decarboxylase in the GABA/glutamine cycle. Evidence that astrocytic pathology may be associated with mood disorders, along with rodent models of mood disorders (see references
15,
16 for reviews), suggests that the abnormalities observed in the amino acid neurotransmitter systems associated with mood disorders could be secondary to changes in the glutamate/glutamine and GABA/glutamine cycles.
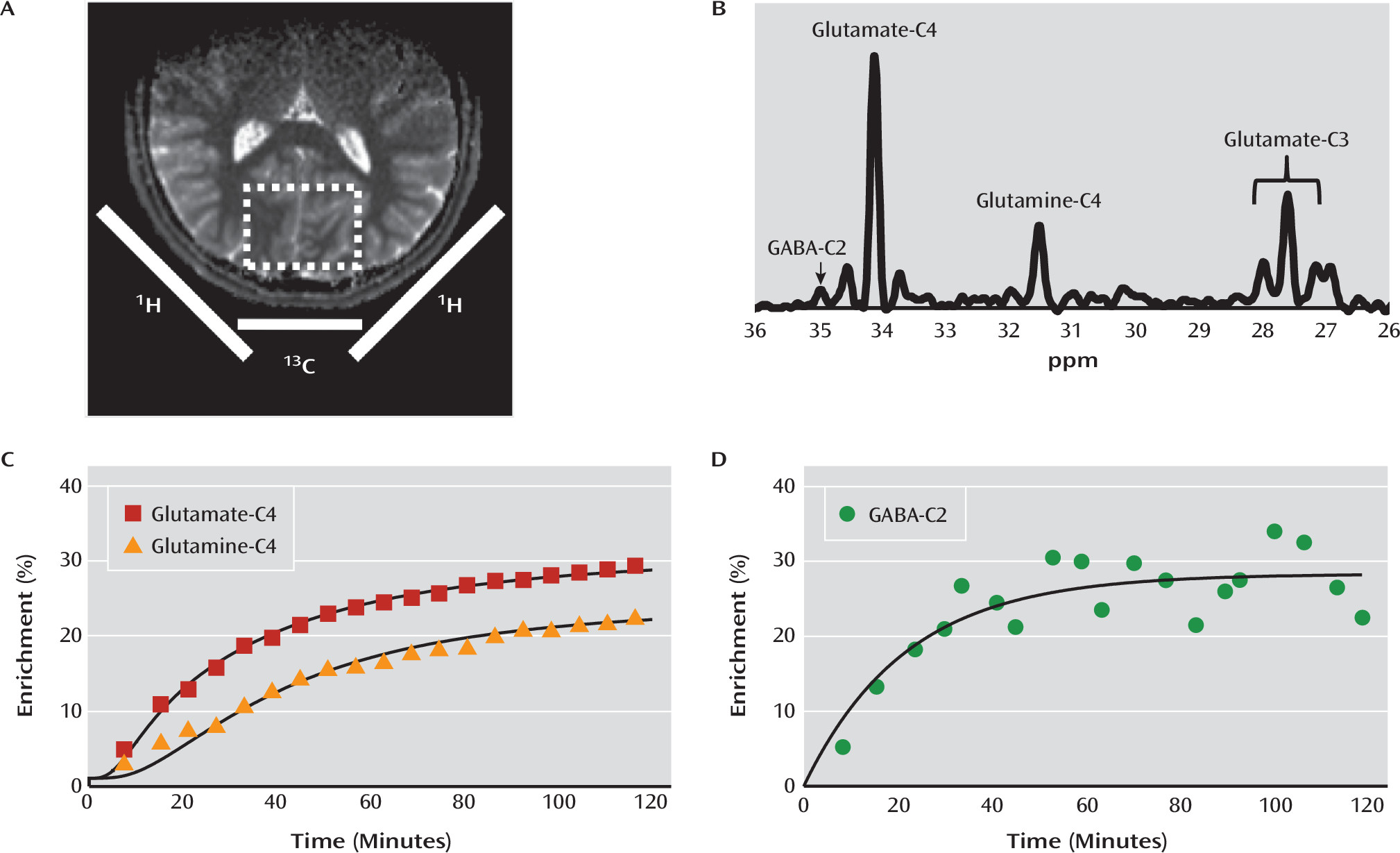
Given the rapidly mounting evidence suggesting that altered mitochondrial function and amino acid metabolism are associated with depression and other mood disorders (
3), we used carbon-13 MRS (
13C MRS) to determine whether the rate of the neuronal TCA cycle (V
TCAN), the glutamate/glutamine cycle (V
cycle), and GABA synthesis (V
GAD) are altered in major depressive disorder. We found a significant reduction in the rate of the neuronal TCA cycle in glutamatergic neurons, implicating the glutamatergic system and mitochondrial energy metabolism as having an important role in the pathology of major depression. Paradoxically, we also found that V
cycle was similar in depressed and healthy comparison subjects, implying a possible alteration in neuronal coupling. To our knowledge, this is the first study to use
13C MRS in vivo to study the pathophysiology of major depression, and it provides the first measures of V
GAD in humans as well as the first correlations between
13C MRS and
1H MRS measures of metabolite concentrations in humans.
Discussion
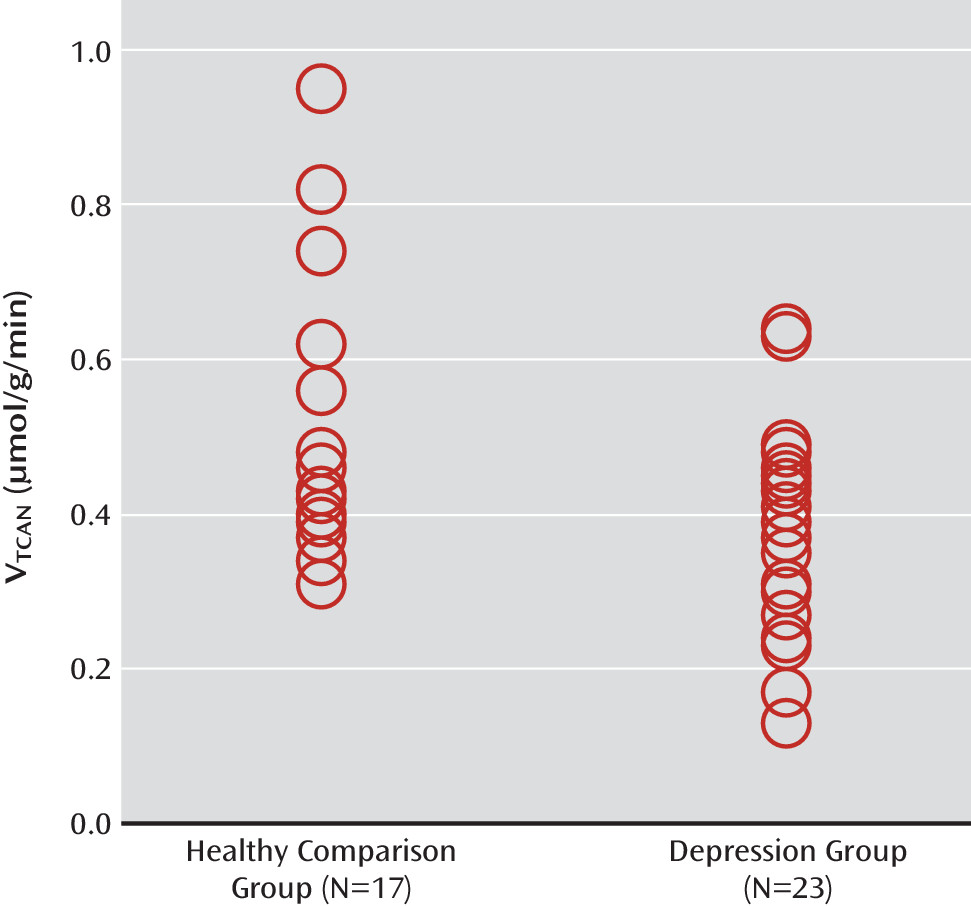
Using state-of-the-art
13C MRS methods, we studied neuronal oxidative energy production, glutamate-glutamine cycling, and GABA synthesis in individuals with major depression and healthy comparison subjects. We found that oxidative energy production specific to glutamatergic neurons was 26% lower in the depression group. No differences between groups were observed in glutamate-glutamine cycling or GABA synthesis. Total glutamate levels were correlated with glutamate-glutamine cycling and, to a lesser extent, neuronal energy production. However, in contrast to findings in previous studies (
8,
9), amino acid neurotransmitters did not differ between groups in this sample. Finally, post hoc exploratory analysis—without correction for multiple comparisons—showed a negative association between glutamate concentration and number of depressive episodes, as well as between glutamine concentration and clinician-rated anxiety scores.
The ability of MRS to separate mitochondrial energy production between glutamatergic neurons, GABA-ergic neurons, and glia is based on the compartmentation of glutamine synthase and glutamic acid decarboxylase in glia and neurons, respectively, and the majority of the neuronal glutamate pool being in glutamatergic neurons (see reference
14 for a review). Because MRS can distinguish carbon-13 labeling in GABA, glutamine, and glutamate, it is possible to use kinetic modeling to obtain separate measures of the TCA cycle in all three compartments (
14,
26,
29).
The findings of reduced neuronal energy production in the depression group are generally consistent with reports of slower energy metabolism, primarily decreased baseline levels of β-nucleoside triphosphate (β-NTP) and total NTP (associated with major depression [
30,
31]), and with reports from positron emission tomography studies of regional and global reductions in glucose metabolism in major depression (
32). In addition, in a rodent model of chronic unpredictable stress, reduced carbon-13 labeling from glucose was observed in glutamate, consistent with findings in human subjects (
33).
A potential explanation for the reduced neuronal energy production is mitochondrial impairment in depressed individuals. Mitochondrial dysfunction in mood disorders has been suggested previously (reviewed in reference
3). However, direct in vivo measures of neuronal mitochondrial energy production in major depression have not previously been investigated. Impaired oxidative metabolism, in the absence of reduced neuronal activity, would be consistent with previous evidence suggesting an increase in cerebral glycolysis and lactate, as well as reductions in phosphocreatine level and pH in patients with mood disorders (
3,
34).
An alternative explanation is that the reduced neuronal energy production reflects a down-regulation of cortical activity. Extensive animal and human studies have demonstrated a strong coupling, close to 1:1, between energy production in the TCA cycle and the glutamate/glutamine cycle (
17). Consistent with this relationship, we observed a positive relationship, approaching statistical significance, between V
TCAN and V
cycle (r=0.436, p=0.08) in the healthy comparison subjects in the present study (see Table S4 in the online
data supplement). In the depression group, however, the relationship between V
TCAN and V
cycle appeared to be weaker (r=0.252, n.s.). This is again reflected in the fact that we observed normal rates of glutamate/glutamine cycling despite slower energy production in the depression group.
The seemingly contradictory findings of reduced neuronal energy metabolism and normal levels of glutamate/glutamine cycling could be reconciled by the presence of an overall reduction of glutamatergic synaptic strength. Brain functional energy needs are largely determined by the ATP needed to maintain ion flows in the post- and presynaptic terminals of excitatory synapses that are coupled to glutamate neurotransmission (
17). Reduced overall synaptic strength in individuals with depression would reduce energy demands for the same amount of glutamate-glutamine cycling. In line with this hypothesis, chronic stress has been shown to reduce glutamate AMPA and NMDA receptor expression and transmission, the primary determinants of synaptic strength (
35).
Alternatively, the rate of the glutamate/glutamine cycle in the depression group may have been overestimated because of a change in the balance between neuronal and astroglial metabolism in this group. A previous
13C MRS study of healthy aging (
22) found that the reduction in neuronal metabolism is accompanied by an increase in glial metabolism, and similar changes in major depression could lead to an overestimate of the rate of the glutamate/glutamine cycle with the kinetic modeling used. Increasing evidence suggests astroglial changes in major depression (
16,
36), including in
13C MRS studies in a rodent model of chronic unpredictable stress (
33). To overcome this limitation, future studies may employ recently developed carbon-13 methods of combined labeling by
13C-glucose and
13C-acetate (
37). Acetate oxidation is limited to astroglial cells, so the simultaneous administration of
13C-glucose and
13C-acetate allows separate measurements of neuronal and astroglial energy production, respectively (
14). In addition, double labeling would enhance the precision of glutamate-glutamine cycling estimates, providing additional insight into the relationship between metabolic fluxes in individuals with major depression compared with healthy subjects.
In addition to the lack of a direct measure of astroglial metabolism, other limitations of this study include its strict criterion of enrolling only individuals who have been medication free for at least 4 weeks in an attempt to minimize any effects of medication withdrawal. This criterion may have affected the characteristics of the depression group, excluding individuals with more treatment-resistant and melancholic depression. This in turn could have contributed to the lack of GABA-ergic differences between groups in contrast to previous studies, where reduced GABA was found primarily in individuals with melancholic and treatment-resistant depression (
8,
9). Although we had a limited number of participants meeting criteria for the melancholic subtype of major depression, we observed a pattern suggesting that they may have had lower GABA levels relative to the healthy comparison subjects and others in the depression group (Figure S1 in the online
data supplement).
Given the focus on the prefrontal cortex in depression, the occipital cortex volume we studied could be considered a major limitation. However, this is the region where significant changes in amino acid neurotransmitter content have previously been reported (
8,
9). Additionally, a growing number of studies have demonstrated abnormal occipital cortex function in individuals with depression. The studies most consistently demonstrate altered levels of occipital cortex activation to emotionally laden visual stimuli (
38,
39). Interestingly, the abnormal stimulus-processing biases seen in patients with depression are reported to normalize with treatment, and a recent study suggests that the magnitude of neural response in the middle occipital cortex may provide a biomarker that predicts response to the rapidly acting antidepressant effects of scopolamine (
40).
In conclusion, the data presented here provide evidence of reduced oxidative energy production within glutamatergic neurons from individuals with major depression. The strengths of this study include the use of 13C MRS in the study of psychiatric disorders to interrogate glutamatergic activity in vivo in humans, as well as the first quantitative measurement of the rate of GABA synthesis in the human cerebral cortex. The reduction in oxidative mitochondrial energy production in glutamatergic neurons could be related to several possible pathophysiological processes, including mitochondrial dysfunction, reduced levels of glutamatergic synaptic activity, and altered coupling of neuronal-astroglial metabolism. Studies employing animal models that specifically examine the relationships between these potential factors and oxidative metabolism will help determine the mechanisms underlying the finding. Future studies identifying the pathophysiological changes underlying the reduced levels of oxidative energy production could provide novel targets for the development of new therapeutics.