Depression, anxiety, and stress affect a dyad during pregnancy—the woman and her future child. Consistent with the Developmental Origins of Health and Disease model (
1), prenatal distress is associated with heightened psychiatric risk (
2), suggesting a “third pathway” for the familial transmission of disease beyond shared genes and the postnatal effects of maternal psychopathology: the influence of pregnant women’s distress on fetal neurobehavioral development. However, the mechanisms underlying this prenatal transmission of risk remain unclear (
3). The placenta, which regulates the prenatal environment, is a focus of emerging research suggesting that maternal distress exerts epigenetic influences on glucocorticoid-related genes and, in turn, infant outcomes (
4). However, no prior studies have considered maternal distress and the hypothalamic-pituitary-adrenal (HPA) axis in relation to placental gene regulation and fetal behavior, which could provide substantiating evidence for the in utero shaping of children’s future mental health.
Children exposed to maternal prenatal distress are at an increased risk for psychopathology (
5), including attention deficit hyperactivity disorder and cognitive deficits (
5), with later life emergence of schizophrenia and affective disorders (
6). Experience-associated alterations in maternal HPA axis, in particular increased cortisol, is a potential mediating pathway (
7), a conclusion reinforced by data from animal models (
8). However, in humans, associations between women’s distress and HPA-axis activation are inconsistent, which has limited the possibility of detecting a biological mediator of the prenatal effects (
7).
Recent findings suggest that heightened maternal anxiety increases the association between plasma and amniotic cortisol levels (
9), and it has been suggested that women’s distress may affect children’s outcomes via epigenetic regulation of glucocorticoid pathway genes in the placenta. Higher anxiety was found to be associated with a downregulation of placental mRNA for the gene encoding 11β–hydroxysteroid dehydrogenase type 2 (
HSD11B2) (11β-HSD-2 is a barrier enzyme that inactivates glucocorticoids [
7]), and higher anxiety and modestly increased DNA methylation of placental
HSD11B2 were together associated with lower muscle tone in newborns (
10). Maternal depression and greater placental DNA methylation of
NR3C1, the gene encoding the glucocorticoid receptor, predicted poorer self-regulation, lower muscle tone, and more lethargy in neonates (
10,
11), although other reports showed maternal adversity (depression, low socioeconomic status) to be associated with increased placental levels of
NR3C1 mRNA (
12,
13). Placental DNA methylation of
FKBP5 (FK506 binding protein, a molecular chaperone of glucocorticoid receptor regulation) was associated with an increased likelihood of high arousal in newborns (
14). In cultured cells, glucocorticoids upregulated placental
HSD11B2 expression (
15); their effect on placental
NR3C1 and
FKBP5 has not been characterized, although cortisol-associated alterations in the functioning of both genes are involved in glucocorticoid resistance and basal hypercortisolemia (
16).
Evidence of maternal prenatal distress and epigenetic changes in the placenta influencing newborn neurobehavior validates the Developmental Origins of Health and Disease model by isolating the effect to the in utero period. Examining fetal neurobehavior provides the same advantages. Neurobehavioral assessment before birth, specifically the cross correlation between fetal movement and heart rate, “coupling,” provides an index of fetal CNS development (
17). Coupling reliably increases over gestation, changing from typical values of 0.40–0.70 (
3,
18,
19), reflecting the coordination of the autonomic and somatic systems (
17), and is positively associated with more mature neural integration at birth based on brain stem auditory-evoked potentials (
20). Maternal cortisol is associated with alteration in the expected coupling increase, and distress is inversely associated with coupling levels (
19).
In the present study, we examine four assessments of second-trimester maternal distress (perceived stress, depression, anxiety, and pregnancy-specific stress), as well as an assay of the HPA axis (daily cortisol) in relation to DNA methylation of three glucocorticoid-related placenta genes (
HSD11B2,
NR3C1, and
FKBP5) and third-trimester fetal coupling. We tested the hypothesis that higher distress would predict increased DNA methylation in these genes and that elevated cortisol would be associated with less DNA methylation of
HSD11B2 yet greater DNA methylation of
NR3C1 and
FKBP5 based on glucocorticoid receptor downregulation by glucocorticoids in other organs and evidence that dexamethasone produces decreases in glucocorticoid receptor mRNA in placenta trophoblast cells (
15). We further predicted that maternal prenatal distress and cortisol would influence fetal neurobehavior and tested whether changes in DNA methylation correlated with these associations.
Method
Participants
Healthy pregnant women, ages 18–45, were recruited through the Department of Obstetrics and Gynecology at Columbia University Medical Center. Women were excluded if they acknowledged smoking or use of recreational drugs, lacked fluency in English, were multiparous, or taking medications. Over a 2-year period (2011–2013), 110 participants were recruited. To allow for replication of our findings, this report describes our targeted gene approach in 64 participants (three participants were excluded from all analyses because they gave birth prior to 37 weeks gestation). One woman conceived using a donor egg. The analyzed and nonanalyzed participants did not differ on variables (age, body mass index [BMI], gestational age at birth, birth weight, infant sex, race/ethnicity, or three out of four mood variables; the unanalyzed sample scored higher on the Pregnancy Distress Scale). All enrolled participants provided written, informed consent, and all procedures were approved by the institutional review board of the New York State Psychiatric Institute/Columbia University Medical Center and Weill Cornell Medical College.
Study Procedures
Sixty-one pregnant women completed mood questionnaires and daily salivary cortisol collection between 24 and 27 gestational weeks and underwent fetal assessment at 34–37 weeks; each of these study sessions involved three consecutive days of testing (see reference
3). At birth, a placenta sample and medical data were collected.
Maternal Characteristics and Birth Outcomes
Gestational age at birth, birth weight, C-section status, and sex of the infant were determined from the medical record. Gestational age at study sessions was determined based on ultrasound examinations and last reported menstrual cycle. BMI was calculated using pre-pregnancy weight from self-report and measured height, along with maternal age.
Salivary Cortisol
Forty-eight hour salivary cortisol collection began during the first day of each study session. Subsequent samples on the second day were collected at waking; at 45 minutes, 2.5 hours, 3.5 hours, and 8 hours after waking; and at 10:00 p.m. or before going to bed. The Medication Event Monitoring System track cap (Aardex, Union City, Calif.) was used to monitor collection times. After collection, cotton was placed in a Salivette tube (Sarstedt, Newton, N.C.), returned to the laboratory, and frozen at −80°C. Cortisol was measured by ultra-performance liquid chromatography-tandem mass spectrometry assay developed by the Irving Institute for Clinical and Translational Research, Columbia University Medical Center. The lower limit of quantitation was 100 pg/mL. Intra-assay and interassay coefficients of variation are less than 3.4% and 3.6% over the analytical measurement range (100 pg/mL–50,000 pg/mL).
Maternal Distress
Participants completed four questionnaires. Two were self-report questionnaires: the Prenatal Distress Questionnaire (
21) and the Perceived Stress Scale (
22). And two were interview-based questionnaires: the Hamilton Depression Rating Scale (HAM-D) (
23) and the Hamilton Anxiety Rating Scale (HAM-A) (
24).
Fetal Assessment
Participants were in a semi-recumbent position for 20 minutes as fetal movement and heart rate were acquired. Data were obtained using a Toitu MT 325 fetal actocardiograph (Toitu Co., Ltd., Tokyo), which detects fetal movement and heart rate via a single transabdominal Doppler transducer. The fetal data were collected from the Toitu’s output port, digitized at 50 Hz using a 16-bit A/D card (National Instruments 16XE50) and analyzed offline; the cross-correlation (“coupling”) of fetal movement and heart rate was calculated as previously described (
3).
Analysis of CpG Methylation in Placentas
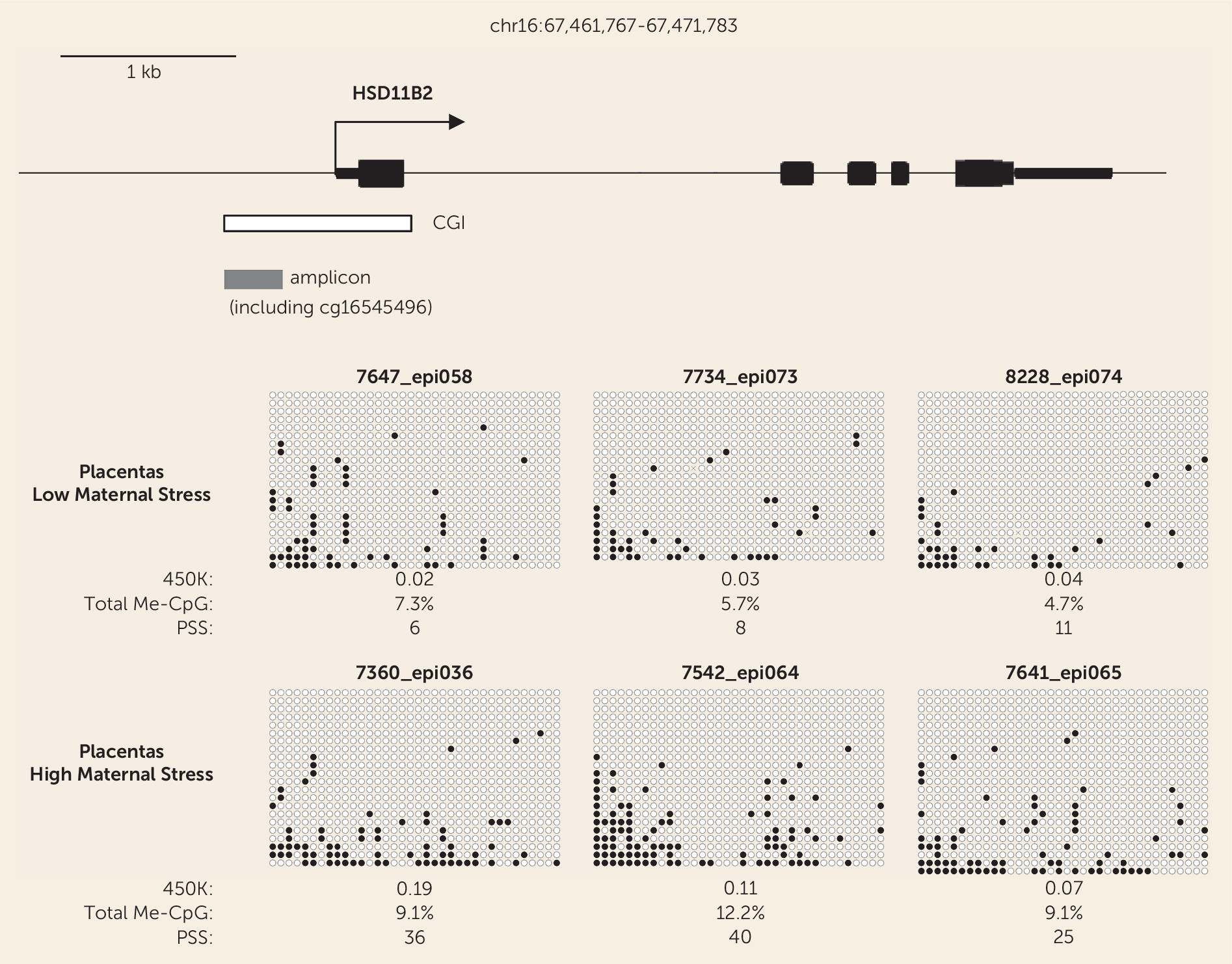
A 1-cm3 sample of the placenta near the fetal surface was collected, and genomic DNA extracted. DNA was assessed by agarose gel electrophoresis with ethidium bromide staining and by PicoGreen assays (Life Technologies, Carlsbad, Calif.). DNA, 500 ng, was bisulfite converted and analyzed according to the manufacturer’s instructions for Illumina HumanMethylation 450K BeadChips, with all assays performed at the Roswell Park Cancer Institute Genomics Shared Resource. Data were processed using Genome Studio software, which calculates the fractional methylation (AVG_Beta) at each CpG, after background correction and normalization to internal controls. All probes querying CpGs that overlapped common single-nucleotide polymorphisms (SNPs) (SNPs with minor allele frequency ≥1% in dbSNP build 138) were removed. Bisulfite sequencing was performed by converting the DNA samples using the EpiTect Bisulfite kit (Qiagen, Hilden, Germany), amplifying the DNA by polymerase chain reaction (PCR), cloning using the TOPO TA Cloning kit (Life Technologies, Carlsbad, Calif.), with three independent PCRs pooled for each sample prior to bacterial transformation, and sequencing of multiple clones.
Data Transformation and Analysis Plan
To index HPA axis activity, cortisol area under the curve was calculated from the wake-up time to bedtime on the second day of collection because the second day included a time-based, uniform protocol (as previously described [
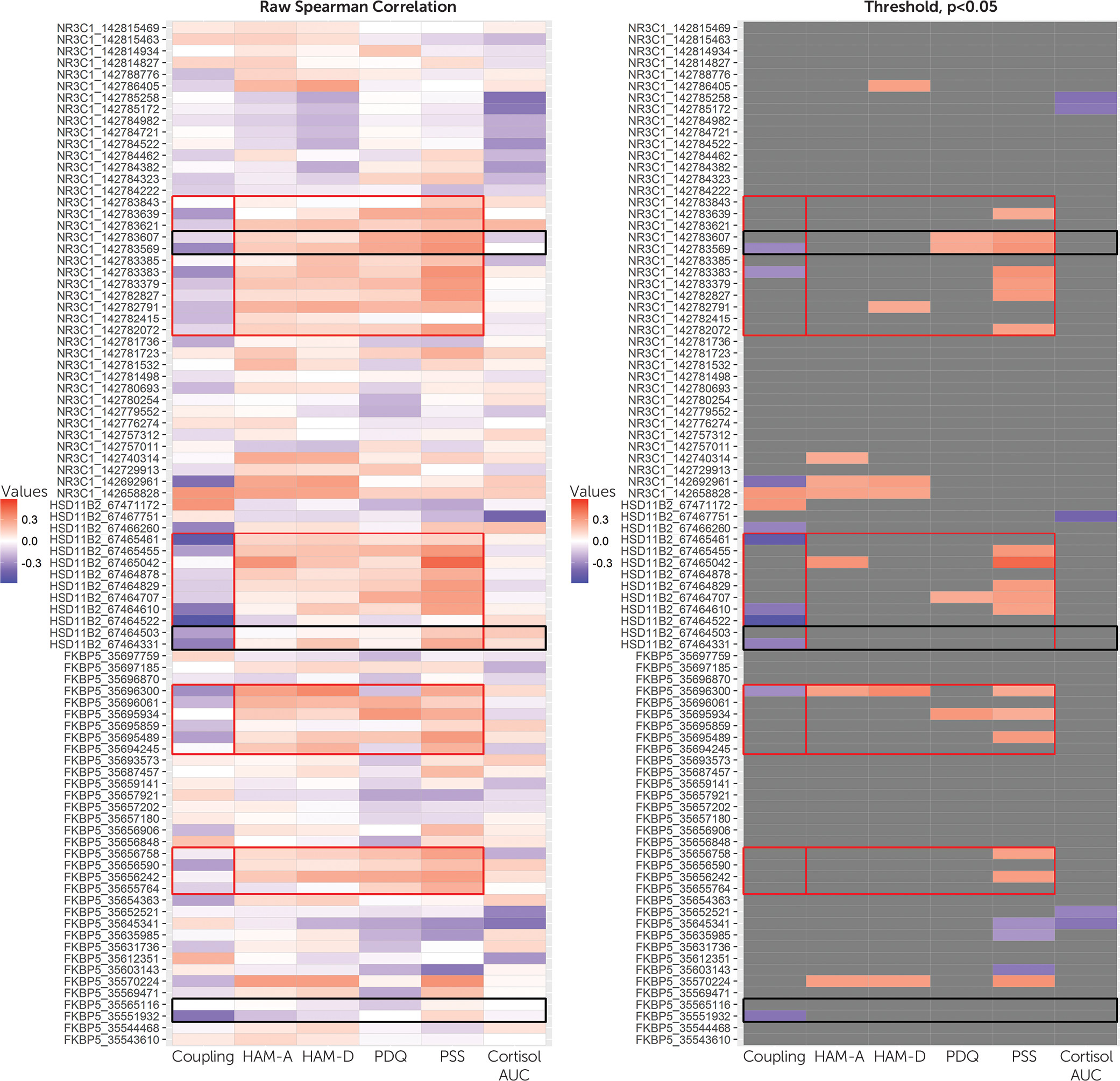
3]). For inclusion in area under the curve analyses, we required the first wake-up collection and ≥8-hour time span from the first to last sample. Cortisol area under the curve data were not normally distributed and were log transformed to satisfy normality by the Kolmogorov-Smirnov test. Descriptive statistics were calculated using mean and standard deviation for continuous variables and percent for categorical variables. Spearman’s rank-order correlation analyses were used to examine associations between DNA methylation outcomes and covariates of interest and to identify bivariate relations between differentially methylated CpG sites within
NR3C1,
HSD11B2, and
FKBP5 and fetal outcomes and maternal variables. We performed mediation analyses to investigate the hypotheses that maternal distress may have an effect on fetal development via DNA methylation. We used structural equation modeling with bootstrapping to estimate and test the significance of direct and indirect effects. In all analyses, infant sex, gestational age-corrected birth weight, and C-section status were included as covariates. Analyses were run including and excluding the donor egg participant with no differences identified.
Discussion
To our knowledge, this is the first study to identify the effects of pregnant women's distress on their offspring in utero and epigenetic changes in placental gene DNA methylation as a potential pathway for this influence. Corroborating prior studies (
11,
29), higher perceived maternal prenatal stress was associated with mildly increased DNA methylation of glucocorticoid-related genes—
HSD11B2,
FKBP5, NR3C1—in the placenta; unique to this report, increased DNA methylation of
HSD11B2 and
FKBP5 was associated with reductions in a key fetal outcome (coupling) predictive of infant neurobehavioral development (
20). Research on developmental origins of health and disease has shown associations between women’s adverse pregnancy experiences and children’s heightened risk for psychopathology (
2), and thus our results here provide proximal evidence for the putative prenatal shaping of children’s mental health trajectories.
Consistent with the low-risk status of our study’s sample, specifically their healthy birth outcomes and low average levels of reported distress, there were small degrees of placental DNA methylation variation for our three targeted genes (
30,
31). However, even in this context, women’s perceived stress was positively associated with higher DNA methylation. Our identified methylation blocks for
HSD11B2 and
NRC31 overlapped with CpG loci in other similar research (
30) (see
Figure 1). With respect to
HSD11B2, our findings are consistent with those of our prior work based on an animal model (
32), as well as other studies with humans (
11,
27,
29), showing that DNA methylation of this gene varies as a function of adversity during pregnancy. Increased DNA methylation of
HSD11B2 leads to a downregulation of associated placental mRNA and its encoded protein, a barrier enzyme that inactivates cortisol (
7). Elevated in utero exposure to cortisol is associated with changes in brain-behavior development, including increased emotionality and impaired learning and motor development (
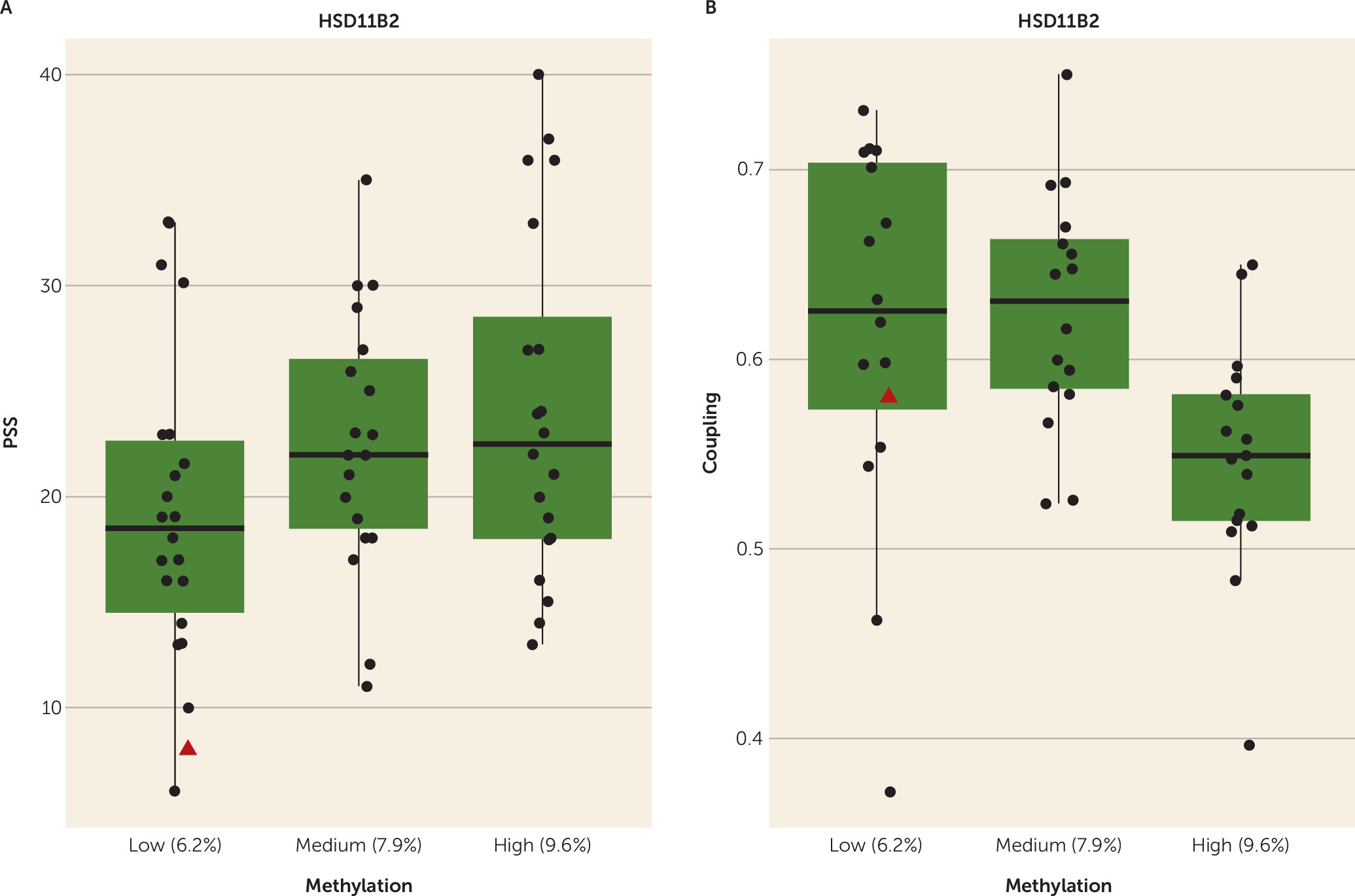
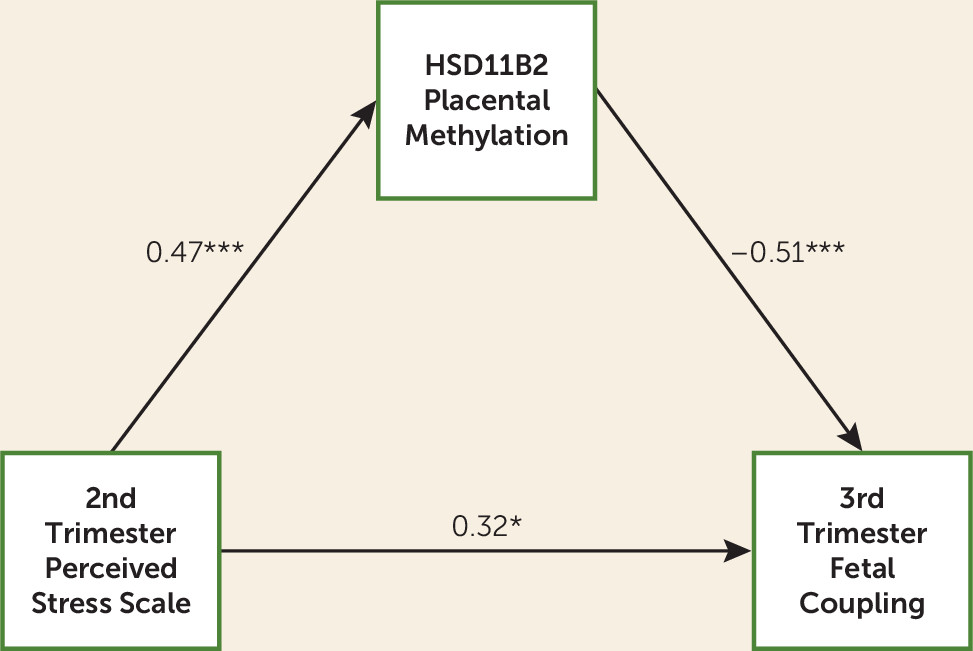
8). Here, higher perceived stress was associated with greater
HSD11B2 DNA methylation, which in turn was associated with reduced fetal coupling. In reports from our group (
3), as well as others (
33), fetal exposure to higher levels of maternal cortisol is associated with less of the expected increase in coupling over gestation (
3) and a failure to respond to a vibro-acoustic stimulus (
33). Fetal coupling reflects CNS development (
17), and higher levels of coupling are associated with more mature neural integration at birth (
20). Taken together, the findings suggest that maternal prenatal distress alters placental regulation of fetal cortisol exposure via increased DNA methylation of
HSD11B2, resulting in a fetal risk phenotype of reduced coupling. However, as in a prior report (
3), after controlling for
HSD11B2 DNA methylation, greater maternal perceived stress had a direct and positive association with fetal coupling level, which is consistent with other data indicating that maternal distress exposure can have a facilitative effect on fetal development (
20) and the possibility that maternal psychological stress influences fetal development independent of HPA axis activation (
34).
Interestingly, our bisulfite sequencing data showed that a majority of placental cells in both high- and low-stress cases have no DNA methylation in the HSD11B2 promoter region. Thus, the biologically relevant epigenetic change due to stress may be affecting only a small subpopulation of placental cells. Whether this subpopulation is specific to a particular layer, such as syncitio- or cytotrophoblast or mesenchyme, or reflects stochastic changes, remains to be defined by future work.
Higher maternal distress also was associated with increased placental DNA methylation of
FKBP5, which in turn predicted reduced fetal coupling. One other report has related higher placental DNA methylation of
FKBP5 to increased arousal in newborns (
14).
FKBP5 decreases the binding of cortisol to its receptor, leading to reduced cortisol responses; thus it may be that increased
FKBP5 DNA methylation leads to greater cortisol activation of glucocorticoid receptor targets within the placenta, leading to overactivation of the HPA axis pathway in the developing fetus (
14). In animal models, manipulations to increase in utero cortisol exposure result in reduced hippocampal weight and synaptogenesis, as well as altered density of glucocorticoid receptors in the amygdala and hippocampus, with consequent decrements in learning and memory (
8); our coupling findings may reflect a downstream early behavioral marker of altered placental glucocorticoid receptor sensitivity.
Contrasting with one report of higher cortisol leading to upregulation of
HSD11B2 expression in cultured cells (
15), yet similar to our results for DNA methylation from infant buccal swabs (
35), we found no consistent associations between maternal cortisol and DNA methylation of glucocorticoid-related genes in the placenta. This could be a consequence of our limited cortisol assessment or our strategy to choose blocks of CpG sites for analyses based on contiguity across CpGs and associations with maternal distress. Inspection of
Figure 1 reveals that individual CpG sites were negatively associated with cortisol levels; we plan to pursue these effects in future studies. Unlike other studies showing associations between
NR3C1 DNA methylation and newborn outcomes (
30,
36), we found no associations with fetal outcomes. Such inconsistencies could be resolved in larger cohorts using common CpG sites and fetal parameters through early development. Finally, we did not detect sex differences in placental DNA methylation, possibly due to our small sample size.
For this sample of generally low-distress pregnant women, an index of greater perceived stress was most consistently associated with increased placental DNA methylation across all three genes of interest. This finding indicates that during pregnancy, even the relatively common life experiences of feeling unable to “control important things in your life” and “cope with all the things you have to do” (
22) are associated with alterations of DNA functioning in the placenta that, in turn, affect fetal development. That depression and stress were highly associated in this sample suggests that depressed pregnant women may influence fetal development via the associated biological changes in the placenta linked to perceived stress and indicate that management of life stress may be an effective intervention strategy.
In human studies of the Developmental Origins of Health and Disease model, it has been difficult to detect a biological mediator of prenatal maternal mood effects on the future child (
7). Here, for the first time, we show a candidate mediator: namely, alteration in placenta DNA methylation. However, as in other reports (
27,
29,
37,
38), we did not identify which biological signals enable maternal distress to affect placental DNA methylation. Genetic variation can significantly alter gene regulation (
10,
14), and it is possible that maternal and/or fetal genes may predispose women to experience stress and influence gene regulation in the placenta, although the finding that the single donor egg subject’s data conformed to our study’s expectations (identified in
Figure 2, as well as Figure S1 in the
online data supplement) offers intriguing evidence against this hypothesis. The brain is socially constructed (
39). Decades of research on developmental origins of health and disease indicate that this process begins in utero. Our data here add molecular support to these findings and underscore the clinical significance of distress during pregnancy as affecting the mother and her future child.