Heavy alcohol use is common in the United States and is associated with multiple adverse consequences (
1). Regular heavy drinking is the major epidemiological risk factor for alcohol use disorder (AUD), a problematic pattern of alcohol use accompanied by clinically significant impairment or distress (
2). Recent genome-wide association studies (GWASs) have identified more than 30 genome-wide-significant variants contributing to the risk of AUD, alcohol dependence, or problematic alcohol use (
3–
5), and more than 100 genome-wide-significant variants have been identified as contributing to alcohol consumption measures (
4,
6,
7) (e.g., score on the consumption subscale of the Alcohol Use Disorders Identification Test [AUDIT-C] [
8], based on the first three questions of the AUDIT [
9]). Nearly all GWAS loci have been identified in individuals of European ancestry, because comparable data from other population groups are not available.
Although the genetic variation contributing to alcohol consumption and AUD partially overlap, multiple variants are associated with one, but not the other, trait (
10). Estimates of the genetic correlation between alcohol consumption and either AUD or problematic alcohol use are moderate (r
g values, 0.52–0.67) (
4,
5), suggesting that despite alcohol use being necessary for AUD, each trait has unique genetic liability. Furthermore, genetic correlations and phenome-wide association studies (PheWASs) of genetic liability for AUD and alcohol consumption show differential patterns of association with physical and mental health (
4–
6).
The genetic divergence between alcohol consumption and AUD may result from confounding, or it may indicate a true difference in the traits’ biology (
11). Potential biases include confounding of alcohol consumption frequency by socioeconomic status (
12) and the heterogeneity of participants reporting alcohol abstinence (
13). However, differences in genetic liability between these traits are observed even after accounting for these confounders. Thus, a latent factor may underlie psychopathology and substance use disorders, but not substance use (
14). As AUD is defined by several diagnostic criteria other than the frequency or quantity of alcohol consumption, individuals can exhibit the same level of alcohol consumption while differing with respect to an AUD diagnosis. Consequently, understanding the factors underlying AUD, separate from those that influence normative alcohol consumption, has important theoretical and clinical implications.
Results
We included 409,630 MVP participants from three ancestral groups: European American (N=296,989), African American (N=80,764), and Hispanic American (N=31,877) (see Table S1 in the online supplement). Most of the population was male (91.5%). Nearly one-quarter (24.7%) of participants had at least one ICD-9 or ICD-10 code for AUD (i.e., AUD–less stringent; the rate was 21.0% among European Americans, 35.2% among African Americans, and 29.2% among Hispanic Americans), and 20.8% had at least one inpatient or two outpatient codes for AUD (i.e., AUD–stringent; the rate was 18.0% among European Americans, 31.0% among African Americans, and 24.6% among Hispanic Americans). The mean number of annual AUDIT-C screenings was 8.1 (SD=3.8; the mean was 8.1 [SD=3.7] among European Americans, 8.5 [SD=3.9] among African Americans, and 7.6 [SD=3.7] among Hispanic Americans). The mean number of AUDIT-C screenings that predated an AUD diagnosis was 3.3 (SD=2.8; the mean was 3.4 [SD=2.9] among European Americans, 3.2 [SD=2.7] among African Americans, and 3.1 [SD=2.6] among Hispanic Americans). The average maximum AUDIT-C score for all individuals was 3.0 (AUD cases, 5.6; control subjects, 2.2) and occurred at an average age of 59.8 years (see Figure S2 in the online supplement).
Identification of Cross-Ancestry and Ancestry-Specific Novel Loci for AUD and AUDIT-C
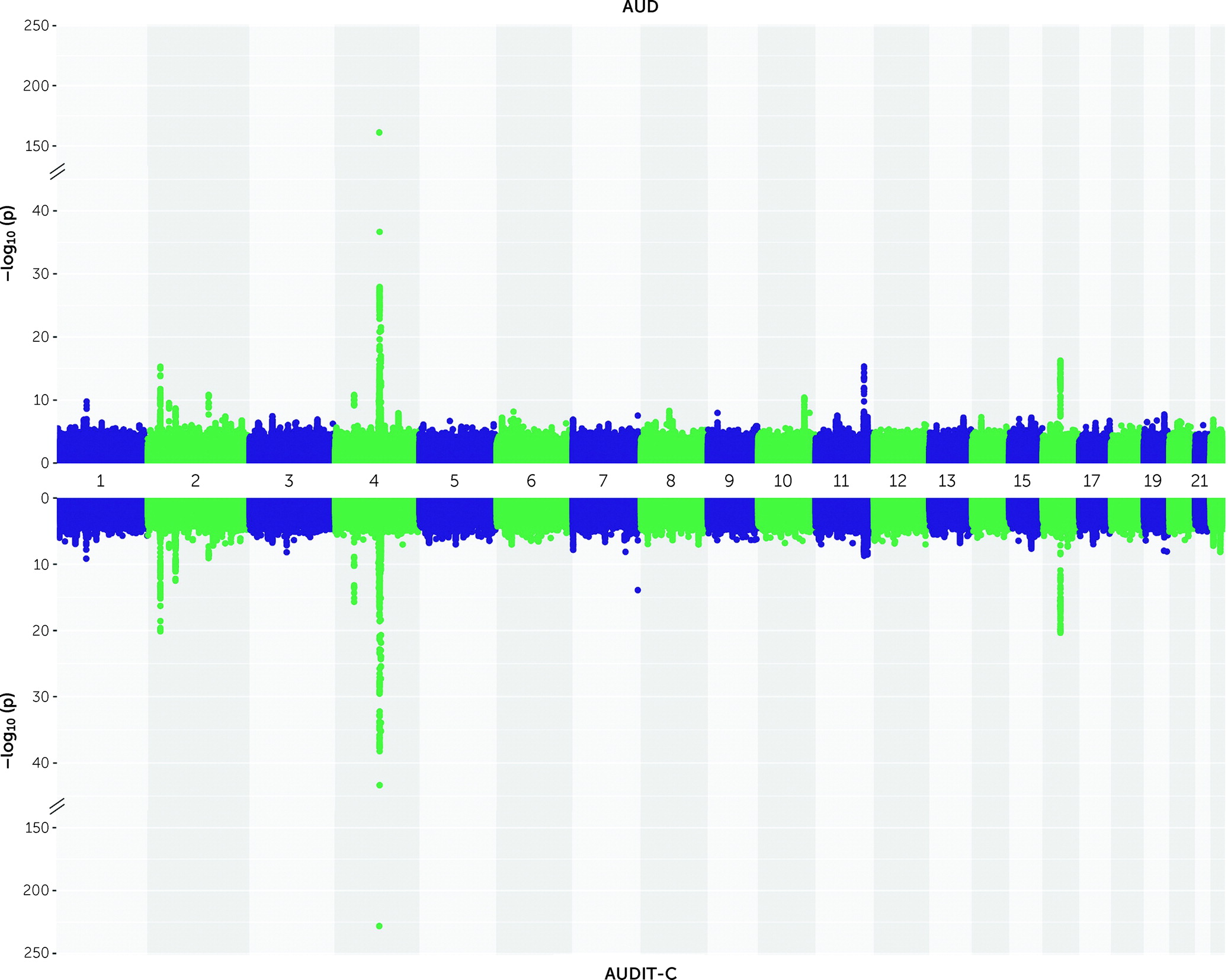
In the cross-ancestry meta-analysis for AUD, 84 lead variants (mapping to 21 loci) were genome-wide significant (p<5×10
−8) (
Figure 1; see also Table S2 and Figure S3 in the
online supplement). After conditioning on the lead variants within each locus, 26 variants were independently associated with AUD, of which four are novel (no previous reported associations with alcohol-related phenotypes within 1 Mb; see Table S4 in the
online supplement), including two intronic (in
ZNF804A and
MLN) and two intergenic (near
NICN1 and
MIR5694). Figure S4 and Table S3 in the
online supplement show the 27 lead variants (14 loci) in European Americans, five lead variants (three loci) in African Americans, and four lead variants (one locus) in Hispanic Americans. Of the lead variants identified in European Americans, four variants in three loci were genome-wide significant in the ancestry-specific analysis only (see Figure S5 in the
online supplement), of which one is novel (near
MIR129-2; see Table S4 in the
online supplement). All European American ancestry–specific variants were polymorphic in all populations (minor allele frequency [MAF] >10%), except rs34517574, which was absent in the African American sample due to low imputation quality. In African Americans, two lead variants in two loci were ancestry specific (see Figure S6 in the
online supplement), with one novel (in an intron of
IL7R; see Table S4 in the
online supplement). Although one of the variants (rs73792114) was rare in European Americans and Hispanic Americans (MAF: African American, 5.9%; European American, 0.04%; Hispanic American, 0.08%), the novel variant (rs7701111) was polymorphic in all populations (MAF: African American, 20.4%; European American, 32.7%; Hispanic American, 42.1%).
In the cross-ancestry meta-analysis for AUDIT-C, 87 lead variants (mapping to 19 loci; see
Figure 1; see also Table S5 and Figure S3 in the
online supplement) were identified. Conditional analysis yielded 24 independent variants, including six novel ones (see Table S7 in the
online supplement). Two of the novel variants are intronic (in
NEGR1 and
CACNG7) and four intergenic (near
GBE1,
LOC101927021,
POT1,
HMG20A). In ancestry-specific analyses (see Figure S7 and Table S6 in the
online supplement), 37 variants (14 loci) were genome-wide significant in European Americans, three variants (two loci) were genome-wide significant in African Americans, and four variants (one locus) were genome-wide significant in Hispanic Americans. In European Americans, two variants in two loci were ancestry specific, with one novel and polymorphic in all populations (see Figure S8 and Table S7 in the
online supplement; near
COBLL1). In African Americans, there was one ancestry-specific variant, which was novel (see Figure S9 and Table S7 in the
online supplement; near
OLFM1); although rare in African Americans, it was almost monomorphic in European Americans and absent in Hispanic Americans (MAF: African American, 0.002%; European American, <0.001%). None of the lead variants in Hispanic Americans were ancestry specific.
We conducted three additional GWASs: 1) AUD in individuals with AUDIT-C score >0; 2) AUDIT-C score in individuals with AUDIT-C score >0; and 3) AUD–less stringent (see Figure S3, Figures S10–S12, and Tables S8–S13 in the
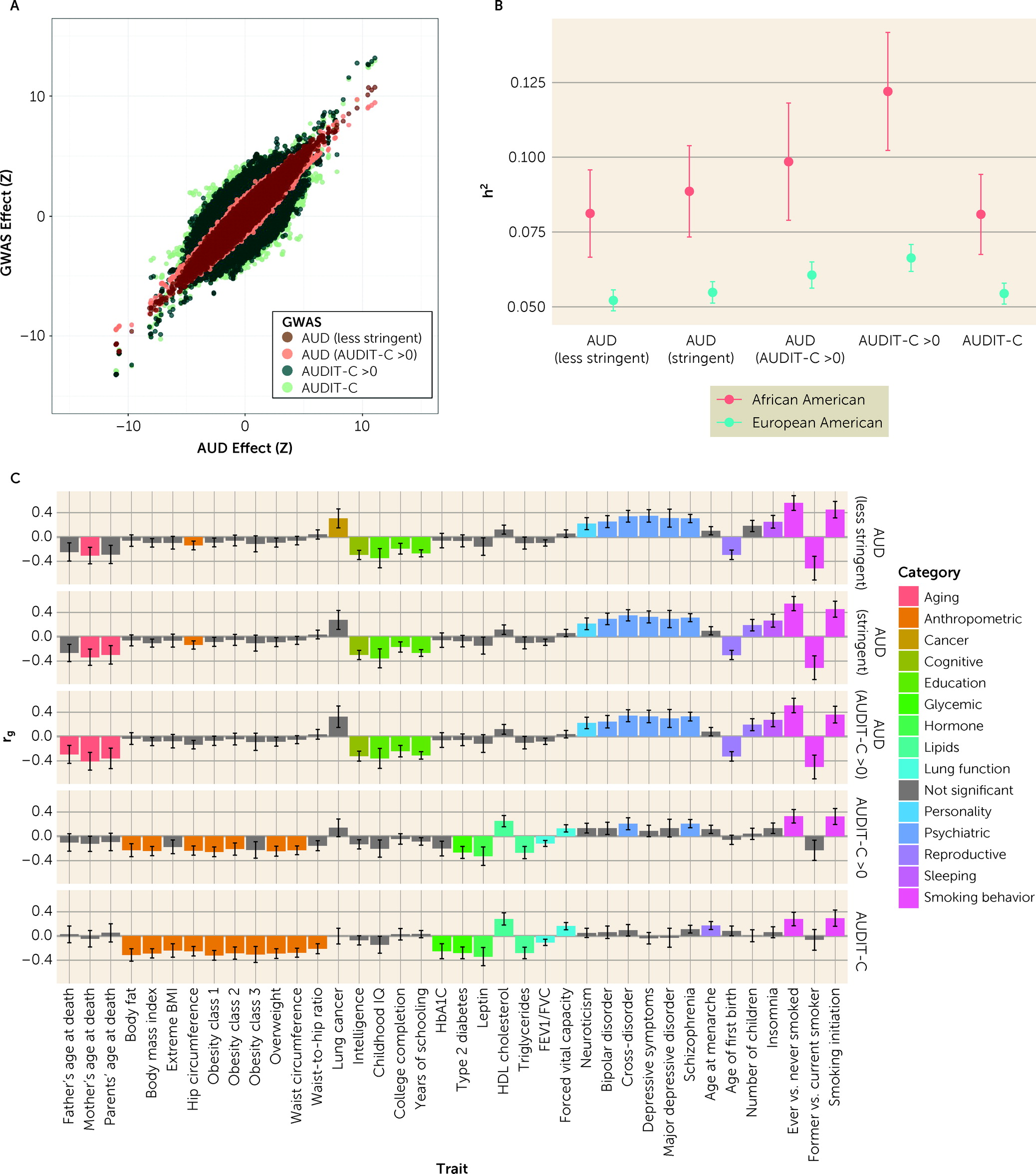
online supplement). In all analyses, correlations between effect sizes for genome-wide significant loci identified in the primary GWAS and the secondary GWAS were high (r
2=0.99). Effect sizes between AUD and AUDIT-C traits were highly correlated (
Figure 2A).
Gene-Based Analyses
Gene-based association analyses for AUD identified 58 genome-wide significant (p<2.69×10–6) genes in European Americans, 10 in African Americans, and seven in Hispanic Americans (see Figure S13 in the online supplement). Genes not identified in the SNP-based analyses of AUD (European American, N=47; African American, N=8; Hispanic American, N=4) include ADH4, which was genome-wide significant in both European Americans and African Americans, ADH5, which was genome-wide significant in African Americans, and ADH7, which was genome-wide significant in Hispanic Americans. Gene-based association analyses for AUDIT-C score identified 45 genome-wide significant genes in European Americans, seven in African Americans, and six in Hispanic Americans (see Figure S14 in the online supplement). In European Americans, 38 of these were not identified in SNP-based analyses, including CACNA1C; six in African Americans, including METAP1; and three in Hispanic Americans, including TRMT10A.
Heritability and Genetic Correlation of AUD and AUDIT-C Phenotypes
SNP-based heritability (h
2SNP) for the five related GWAS phenotypes was estimated using LDSC for both European Americans and African Americans (
Figure 2B; see also Table S14 in the
online supplement). Heritability values, which were higher for African Americans than for European Americans and similar to previous analyses in MVP participants using smaller samples (
4), increased with the specificity of the phenotype. Pairwise genetic correlations (r
g) between the five GWAS phenotypes were high within both European Americans and African Americans (see Figure S15 in the
online supplement). Correlations were strongest within the AUD (r
g range, 0.92–1) and AUDIT-C (r
g range, 0.92–1) traits and weakest between AUD and AUDIT-C (European American r
g range, 0.71–0.78; African American r
g range, 0.93–0.97), but increased when individuals with AUDIT-C scores of 0 were removed (European American r
g range, 0.86–0.92, African American r
g range, 0.98–1). Genetic correlations between European Americans and African Americans within traits were estimated using POPCORN (r
g range, 0.57–0.74; see Table S15 in the
online supplement).
Genetic Correlations With Other Phenotypes
The genetic correlations between the five GWAS phenotypes and 257 other phenotypes were estimated in the European American population using LDSC (
Figure 2C; see also Table S16 in the
online supplement). As in our previous work (
4), the magnitude and direction of genetic correlations differed significantly between AUD and AUDIT-C score (see Table S17 in the
online supplement). AUD was correlated with 22 phenotypes, including positive correlations with several psychiatric disorders, insomnia, and neuroticism, and negative correlations with years of schooling and age at first birth. In contrast, AUDIT-C score was correlated with 20 phenotypes, including positive correlations with HDL cholesterol and lung function and negative correlations with anthropometric phenotypes and type 2 diabetes. Phenotypes significantly genetically correlated with AUD in individuals with AUDIT-C score >0 and AUD–less stringent were generally the same as those correlated with AUD, with no significant differences in magnitude of correlation. Fifteen of the phenotypes that were significantly genetically correlated with AUDIT-C score were also correlated with AUDIT-C score in individuals with AUDIT-C score >0, with no significant differences in magnitude. However, AUDIT-C score in individuals with AUDIT-C score >0 was also significantly positively correlated with cross-disorder and schizophrenia phenotypes, and these correlations were not significantly different from those found for AUD.
Polygenic Risk Scores
In the UK Biobank, PRSs for AUD and AUDIT-C score were significantly associated with scores on the AUDIT-C, the AUDIT-P (the problem items in AUDIT), and the AUDIT in individuals of European ancestry, but not African ancestry (see Table S18 in the online supplement). In BioVU, both PRSs were associated with alcohol-related disorders among individuals of African and European ancestry (see Tables S19–S22 in the online supplement). Among individuals of European ancestry, PheWAS analyses across 1,335 phenotypes identified an additional 35 phenotypes associated with AUD PRS, including positive associations with “tobacco use disorder” and “mood disorders,” and 42 phenotypes associated with AUDIT-C PRS, including negative associations with “type 2 diabetes” and “obesity” (see Tables S21 and S22 and Figure S16 in the online supplement).
Mediation
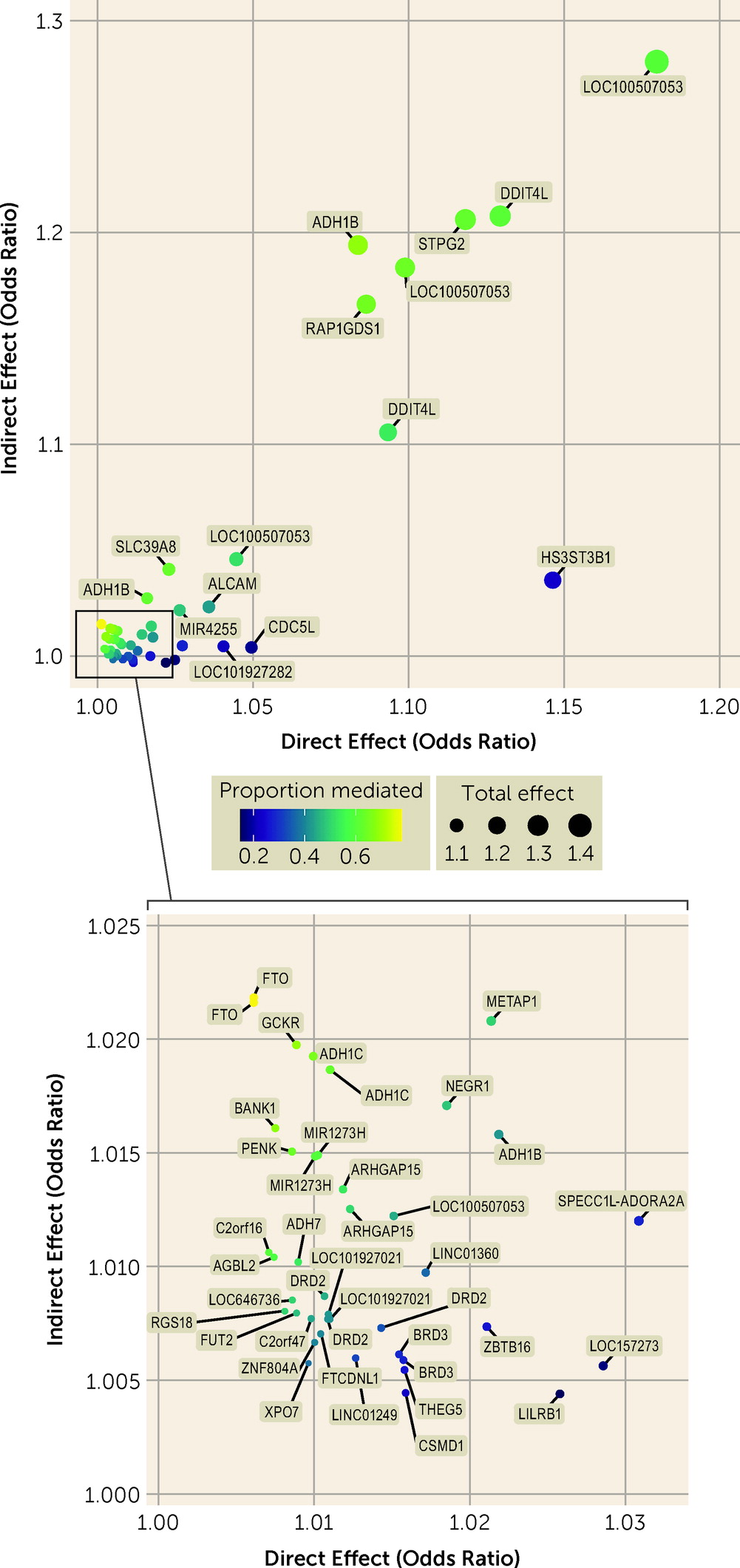
Mediation analysis identified total effects of variants on AUD and AUDIT-C score and direct and indirect effects of variants on AUD (
Figure 3; see also Tables S23–S27 in the
online supplement). In the cross-ancestry meta-analysis, 55 variants were associated with AUD at a Bonferroni-corrected p threshold of 1.34×10
−4, all with a concordant direction of effect on AUDIT-C score. Twelve of these showed no evidence of mediation (i.e., p values >0.05 for an indirect effect and small values for the proportion of mediation), indicating that AUD susceptibility at these loci was not driven by a genetic predisposition for alcohol consumption. These included variants near
LILRB1, LOC157273,
CDC5L,
LOC101927282,
HS3ST3B1,
THEG5,
BRD3, and
LINC01249 and variants in
CSMD1,
ZBTB16, and
SPECC1L-ADORA2A.
Forty-three variants showed evidence for mediation by alcohol consumption on their effect on AUD. The strongest evidence for mediation was found for two variants in FTO (in both cases with odds ratiodirect=1.006, pdirect=0.037; odds ratioindirect=1.022, pindirect=1.59×10−13 and 2.70×10−13), followed by a variant near GCKR (odds ratiodirect=1.009, pdirect=0.002; odds ratioindirect=1.020, pindirect=1.05×10−11). The well-known variant rs1229984 in ADH1B showed both a strong direct effect (odds ratiodirect=1.08, pdirect=2.30×10−26) and a strong indirect effect (odds ratioindirect=1.19, pindirect=4.45×10−117) on AUD, with an indirect effect higher than that for the association with AUD (odds ratio=1.14). This variant also showed evidence for an exposure-by-mediator interaction in European Americans (b=−0.03, p=0.025; see Table S27 in the online supplement), reflecting a decreasing effect of the SNP on AUD as the level of alcohol consumption increases. Other variants with both strong direct and indirect effects on AUD included those near RAP1GDS1 and in SLC39A8.
Discussion
In this study, we performed cross-ancestry GWASs of AUD and AUDIT-C score in 409,630 individuals, including the largest sample of non-European-ancestry individuals for these traits to date. These populations were 40%–50% larger than in our previous GWAS for AUDIT-C score (
4) and AUD (
4), and we identified novel loci in both cross-ancestry meta-analysis and the ancestry-specific analyses. We conducted mediation analysis to establish a set of variants directly associated with AUD, independent of alcohol consumption. We propose that these variants are high-priority candidates for in vitro study of the biology of the transition from alcohol use to AUD, potentially helping to account for differences in risk of AUD among individuals with the same level of alcohol consumption.
We identified 26 loci associated with AUD, including four novel loci in the cross-ancestry meta-analysis and two novel loci in ancestry-specific analyses. Many of the loci, although novel for alcohol-related phenotypes, have been associated with psychiatric disorders or drug use or use disorders. One novel intronic variant in
ZNF804A (Zinc Finger Protein 804A) is located in intron 2, close to a variant previously associated with increased susceptibility to a broad psychosis phenotype (including schizophrenia and bipolar disorder) and heroin addiction (
28). The second novel intronic variant is located in
MLN (Motilin), previously associated with smoking (
7) and depression (
29). A novel intergenic variant located upstream of
NICN1 (Nicolin 1), which has been associated with depression (
30) and intelligence (
31), is a known expression quantitative trait locus for multiple other genes (
32). The novel African American–specific variant is located in the intron of
IL7R (Interleukin 7 Receptor), which plays a critical role in the development of immune cells (
33). Chronic alcohol consumption is known to impair immune responses, and concentrations of IL-7 are altered during detoxification (
34).
Twenty-two loci were associated with AUDIT-C score, including six novel loci in the cross-ancestry meta-analysis and two novel variants in the ancestry-specific analyses. No prior associations with psychiatric disorders have been identified for these novel loci, consistent with the observed divergence in the genetic architectures of AUDIT-C and AUD (
10). Both novel intronic variants are located in genes with prior associations with intelligence and educational attainment (in
NEGR1 [Neuronal Growth Regulator 1] and
CACNG7 [Calcium Voltage-Gated Channel Auxiliary Subunit Gamma 7]) (
31,
35). The novel European American–specific variant near
COBLL1 has been associated with multiple phenotypes, including type 2 diabetes (
36), whereas variants located nearby have shown associations with HDL cholesterol and triglyceride measures in current drinkers (
37). The novel African American–specific variant is located near
OLFM1, a gene associated with smoking behavior (
7).
We refined both the AUD and AUDIT-C phenotypes by removing individuals who never reported alcohol consumption in the VA EHR. In the UK Biobank and the Veterans Aging Cohort Study (
13,
38), where participants’ specific reasons for discontinuing drinking was available, the group not reporting alcohol consumption is heterogeneous, consisting of both lifelong abstainers and former drinkers, some who may have stopped drinking because of health problems, thereby confounding this variable. Confirming this, our analyses demonstrate that 15% of abstainers have an AUD diagnosis in their EHR. Removing them from the analyses did not increase the number of genome-wide significant loci, likely in part because this group makes up almost one-quarter of our original sample. However, by refining the phenotypes to include only individuals who report alcohol consumption, we observed an increase in the SNP heritability for both AUD and AUDIT-C score, and a stronger genetic correlation between AUD and AUDIT-C score. Furthermore, in genetic correlation analyses with other phenotypes, the magnitude of differences between AUD and AUDIT-C score was reduced, revealing positive correlations of AUDIT-C score with psychiatric phenotypes. These findings confirm previous results (
12,
13) in which confounding in measures of alcohol consumption contributed in part to the difference in genetic architecture seen between alcohol consumption and AUD.
Evaluating the mediating effect of AUDIT-C score on the genetic risk of AUD identified variants in three genes involved in alcohol metabolism (alcohol dehydrogenase 1B [
ADH1B], 1C [
ADH1C], and 7 [
ADH7]) with both direct and indirect effects on AUD risk. Variants in these genes alter the rate of alcohol metabolism and are consistently associated with protective effects against excessive alcohol consumption, thereby reducing AUD risk (
39). The direct (nonmediated) effect of these variants on AUD could result from their effects on alcohol consumption earlier in life, prior to the AUDIT-C assessment, or convey risk of AUD via an independent pathway. Another variant with both strong direct and indirect effects on AUD was in
SLC39A8 (solute carrier family 39 member 8; rs13107325), which has been associated with over 60 phenotypes, including anthropometric phenotypes, such as body mass index (BMI) (
40), and psychiatric phenotypes, such as schizophrenia (
41). Thus, this variant may affect risk for AUD both via alcohol consumption and via effects on the psychopathological dimension of addiction.
The effect of variants in
FTO,
GCKR, and
BANK1 on AUD were shown to be largely mediated by their effect on alcohol consumption.
FTO has multiple prior associations with obesity and BMI, with studies suggesting that the mechanism of effect could be through food intake and preference (
42). Likewise, both
GCKR and
BANK1 have prior associations with anthropometric and metabolic phenotypes (
43,
44). Thus, their association with AUD may be due to metabolic effects via increased alcohol consumption.
We identified 12 variants with no evidence of mediation through alcohol consumption on AUD risk. We hypothesize that these variants contribute to an addictive dimension of AUD that is not directly influenced by levels of consumption. Some of these were in or near genes previously associated with schizophrenia (
CSMD1 [CUB And Sushi Multiple Domains 1]) (
41), chronotype (
LINC01249 [Long Intergenic Non-Protein Coding RNA 1249]) (
45), lung function (
CDC5L [Cell Division Cycle 5 Like]) (
46), and caffeine consumption (
SPECC1L-ADORA2A [Readthrough (NMD Candidate)]) (
47). These variants warrant further examination as potential contributors to the transition from heavy drinking to AUD.
Our study has several limitations. First, our measure of alcohol consumption, the AUDIT-C, was self-reported and is subject to recall bias. Second, we systematically captured consumption reported in the VA health system only since 2007; if an individual’s maximum alcohol consumption occurred before then, this measure may not be accurate. Third, different responses to the three AUDIT-C items could result in the same overall score, yet each item may be differentially associated with AUD. In particular, the first item (frequency) is subject to bias and confounding (
12). Future studies could examine each AUDIT-C item separately. Finally, the MVP sample is mostly male (>90%), precluding sex-specific analyses. As AUDIT-C scores vary by sex, this will be an important direction for future studies.
In summary, our study identifies novel associations for AUD and AUDIT-C score, including novel ancestry-specific associations. We addressed some of the differences between the genetic architectures of the traits by reducing heterogeneity in the data set. Although genetic variants that contribute to alcohol consumption are important in our understanding of AUD, there is unique variance that remains after refinement of the two traits. Further, we identified a set of variants with direct effects on AUD that are not mediated through alcohol consumption. We propose these as targets for research aimed at understanding the transition from heavy alcohol consumption to AUD, elucidation of the biology of which could inform prevention and treatment efforts.