Frequent sequelae of traumatic brain injury (TBI), besides neuropsychological deficits such as attention deficits and memory impairment, are sleep disturbances. In medium-term follow-up of young patients after TBI, the most common sleep disturbances were a decrease in the amount of rapid-eye-movement (REM) sleep,
1 a decrease in non-REM sleep stage 1,
2 and an increase in the number of awakenings, resulting in reduced sleep efficiency.
2 Neurobehavioral impairments were more common in patients after TBI with sleep complaints than in those who did not report sleep disturbances, and occupational outcome was poorer.
3In addition, dysregulations of the hypothalamic-pituitary-adrenocortical (HPA) and -somatotropic (HPS) systems have been reported. In an acute intensive care situation, plasma cortisol levels were elevated.
4 Furthermore, patients followed for 2–10 months after TBI frequently showed hypercortisolism at 08:00 h (>20 μg/dl), absence of diurnal cortisol variation, and nonsuppressed cortisol levels after a dexamethasone suppression test.
5 An important role in mediating the intracranial hypertension-induced HPA response after TBI has been suggested for increased arginine vasopressin (AVP).
6 Both corticotropin-releasing hormone (CRH)-secreting and AVP-secreting neurons are located in the paraventricular nucleus around the third ventricle, and this ventricle is often distorted when intracranial pressure is acutely elevated after TBI.
6 With regard to the HPS system, previous investigations have yielded inconsistent results. Whereas after administration of growth hormone (GH)–releasing hormone (GHRH) in TBI patients with a fatal outcome there was no GH stimulation,
7 after glucose loading in TBI patients in an acute intensive care situation there was a significant increase in serum GH.
8 Prolactin (PRL) secretion was increased either soon after TBI, before initial treatment, or more than 6 months after severe TBI.
9 Hyperprolactinemia associated with TBI seems to result from disturbances of hypothalamic neuroendocrine mechanisms that control PRL secretion.
10 Beyond this, dysfunctions of thyroidal and gonadal activity, of glucagon/glucose metabolism, and of antidiuretic hormone secretion have also been reported.
6These findings show that both sleep complaints and alterations of hormone secretion are well known after TBI, even many months later. Studies in patients with psychiatric disorders and control subjects as well as animal research support the notion of a bidirectional interaction between sleep and nocturnal hormone secretion. Associated with the sleep EEG in humans are distinct endocrine patterns, including a rise in plasma cortisol during the second half of the night and GH surges close to the time of sleep onset.
11 CRH and GHRH seem to be reciprocally interacting substances in sleep-endocrine regulation.
12,13 In young male control subjects, CRH administration led to a decrease in slow wave sleep (SWS) and GH secretion and an increase in cortisol secretion,
14 whereas GHRH did just the opposite.
15 There is evidence that the known shift in the GHRH-to-CRH ratio in favor of CRH in depression contributes to abnormal nocturnal hormone secretion.
13,16 In particular, a decrease in SWS, disturbed sleep continuity, disinhibition of REM sleep,
17 elevated sleep-associated adrenocorticotropic hormone
18 and cortisol levels,
13,18 and blunted nocturnal GH release
19,20 are frequent symptoms of depression. The nocturnal PRL secretion, however, is not altered markedly in patients with depression,
21 although regulation of REM sleep and of PRL secretion seem to be linked.
22 In the follow-up after recovery from acute depression and discontinuation of pharmacological treatment, sleep alterations and blunted GH secretion persisted, whereas the acutely increased cortisol secretion normalized.
19,23METHODS
Thirteen brain-injured male inpatients ages 19–36 years (mean=27.3, SD=6.2) were examined in the sleep laboratory of the Max Planck Institute of Psychiatry while under treatment at the Department of Neuropsychology, Munich Bogenhausen City Hospital. The mean interval between TBI and the sleep investigation was 15.5 months (range 2.5–46; SD=12.4). The subjects had had closed severe head injury as assessed with the Glasgow Coma Scale.
26 Four of the patients had been treated with dexamethasone or methylprednisolone in an acute intensive care situation because of elevated intracranial pressure. Five patients had not received steroids after TBI. For the remaining 4 patients, no information was available about medication in the acute situation. At the initial study evaluation, patients were admitted to the trial after passing a medical examination designed to exclude acute illness that could cause ambiguous results. Additionally, patients with local contusional or hemorrhagal brain damage were excluded after magnetic resonance imaging, whereas patients with signs of a diffuse axonal injury like small cerebral marrow hyperintensities or ventricle enlargement were included. None of the subjects had taken pharmaceutical products in the previous 3 months, and none had abused either alcohol or drugs since the accident. The patients had not suffered from depression during the interval between the injury and the study, as assessed by two of the authors (U.M. and D.Y.v.C.). In addition, psychiatric disorders including alcohol and drug abuse and endocrinological disorders in the premorbid lifetime were excluded. At the time of evaluation, none of the patients met the criteria for major depression. On the Hamilton Rating Scale for Depression (Ham-D, 21-item version
27), the total scores were between 2 and 13 points, with 9 patients scoring under 4 points and 4 patients scoring between 5 and 13 points due to somatic and vegetative symptoms (mean=5.6, SD=3.8).
The control group consisted of 13 healthy male paid volunteers with a mean age of 27.1 years (range 22–37; SD=4.4). The subjects were interviewed by a senior psychiatrist to exclude a history of TBI, a personal or family history of psychiatric disorder, and recent stressful life events. Shift workers and subjects who had made a transmeridian flight in the past 6 months were not admitted to the study. Lifetime abuse of alcohol or drugs and use of any medication in the past several months were ruled out. The subjects underwent a physical examination and laboratory tests, including hematology, virology, clinical chemistry, endocrine evaluation, EEG, and ECG.
The protocol was approved by the Ethics Committee for Human Experiments of the Max Planck Institute of Psychiatry, and written informed consent was obtained from all subjects. The first night of each session served for adaptation to the laboratory setting, including application of electrodes and insertion of an indwelling catheter into a forearm vein at 19:30 h. The catheter was connected to a plastic tube that ran through a soundproof lock into the adjacent room. There polygraphic recordings (EEG, electrooculogram, electromyogram, and ECG) obtained in accordance with standard procedure
28 were monitored. The subjects were not allowed to sleep until the lights were turned off at 23:00 h. On the second night of each session, beginning at 22:00 h, blood was drawn every 20 min until 07:00 h through the long catheter. The sleep EEG was recorded between 23:00 and 07:00 h. The polygraphic sleep-EEG recordings were scored visually in 30-second epochs as described in detail previously.
14 Plasma concentrations of the hormones cortisol, GH, and prolactin were determined by standard commercially available radioimmunoassays, which are routinely controlled in our laboratory for batch-to-batch variability and precision. Endocrine variables were calculated as described elsewhere.
14 All group values are expressed as means±SD.
Mean differences between the control and patient groups were tested for significance with the paired Wilcoxon rank test (two-tailed). An alpha value of 0.05 was accepted as the nominal level of significance. Normally the differences had to be tested at a reduced level of significance to keep the type I error less than or equal to the nominal level. However, the small sample size and the choice of the nonparametric Wilcoxon rank test make testing of null hypotheses against the alternative hypotheses very conservative. Supplementary reduction of the nominal level by applying, for example, the Bonferroni correction would make the testing extremely conservative, and that would consequently lead almost surely to nonrejection of all null hypotheses, even in cases where the group means differ strongly from each other.
DISCUSSION
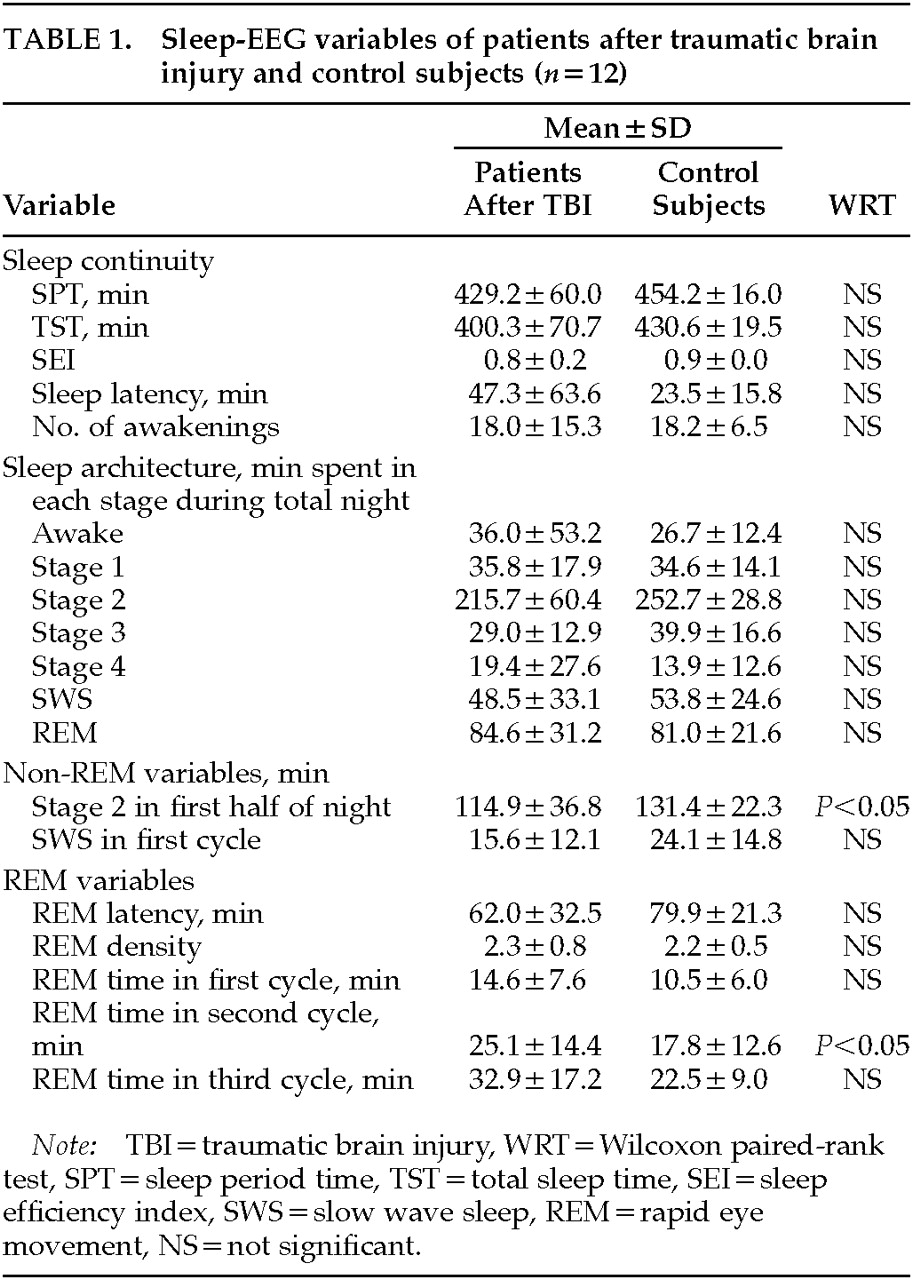
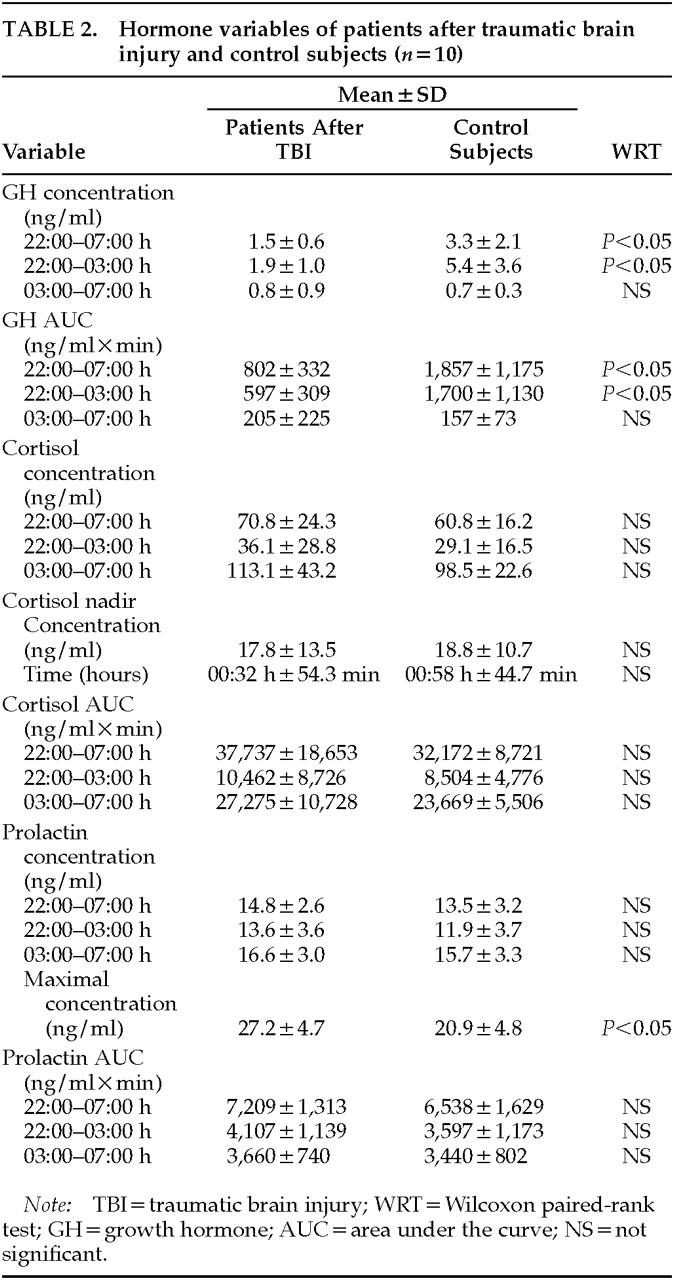
The main finding of the present study is that there are sleep-endocrine alterations several months to several years after TBI. Non-REM stage 2 sleep was reduced in the first half of the night and REM sleep was increased in the second sleep cycle. Furthermore, there was a nonsignificant trend toward REM disinhibition as REM latency was shortened and REM time was increased in the first and third sleep cycles. The nocturnal GH peak was significantly decreased and the maximal PRL secretion was increased, whereas there was no change in cortisol secretion.
The changes in the sleep-EEG variables after TBI in our study are at variance with the results of previous investigations. Ron et al.
1 reported a decrease in REM sleep after TBI, whereas we found a trend toward REM sleep disinhibition. Furthermore, Prigatano et al.
2 found a decrease in sleep efficiency because of frequent awakenings. In our sample, sleep efficiency remained unchanged, but sleep continuity changed because of a reduction of stage 2 sleep and decreases in sleep period time and total sleep time (nonsignificant trend). These discrepancies may have resulted from differences in patient selection. In the study by Ron et al.,
1 all but two of the patients had local damage to brain structures, partly after penetrating injuries. The patients investigated by Prigatano et al.
2 were not drug-free. In contrast, the patients in our sample had not had local brain damage, nor had they taken medication in the previous few weeks. Hence, in our study two major factors that might render sleep-EEG changes ambiguous were excluded through patient selection.
Interestingly, there are similarities between the sleep-EEGs in our sample and in patients with depression. Changes in non-REM sleep in our study were the decrease in stage 2 sleep during the first half of the night and a trend toward reduced SWS in the first sleep cycle. A reduction in the amount of SWS is a frequent symptom in patients with depression. Furthermore, REM sleep disinhibition, which appeared to be present in our sample, is a robust finding in patients with depression.
17 These sleep-EEG changes are not state-dependent. They occur during the acute episode of depression and persist after recovery,
19,29 even for several years.
30 Hence, it appears that impairment of non-REM sleep and disinhibition of REM sleep may be symptoms common to patients with an acute episode of depression or who have recovered from depression and patients who have survived severe TBI.
Furthermore, the pattern of sleep-associated hormone release in the patients after TBI in the present study is similar to that of patients who have recovered from depression. In the TBI patients, GH secretion was decreased, whereas cortisol and prolactin secretion were widely the same as in the control subjects; the only change was an increase of prolactin maximum. The decrease in GH secretion in our study is compatible with observations of a reduced GH stimulation after GHRH administration in an acute intensive care situation after TBI.
7 It is consistently reported that hypercortisolism is a frequent symptom during acute episodes of depression.
31 Accordingly, nocturnal cortisol secretion is enhanced and cortisol nadir is advanced,
18,19 whereas nocturnal GH secretion is blunted in most
19,32 but not all
33 studies. Prolactin secretion in acute depression remains unchanged.
21 In patients with remitted depression, normalized HPA hormone secretion,
18,19,31 but a persisting decrease in GH secretion,
19,20 has been reported. Concomitantly, prolactin secretion shows no abnormalities. In summary, then, there are similarities between the changes in patients after TBI and the changes in patients who have recovered from an acute episode of depression with regard to both sleep-EEG and nocturnal endocrine variables.
We hypothesized previously that the persistence of sleep-endocrine changes in patients who have recovered from depression might represent a biological “scar” resulting from the metabolic aberrations during the acute illness.
19 Alternatively, the abnormalities in patients with remitted affective disorders may correspond to a trait, which should then be found also in premorbid subjects. For example, in healthy humans who have no diagnosis of psychiatric disorder but have a first-degree relative with depression or bipolar disorder, the amount of SWS in the first sleep cycle is reduced and the REM sleep density is increased in comparison to control subjects without a family history of psychiatric disorders.
34 These changes in polysomnography may indicate vulnerability to affective disorders. In brain-injured patients, it appears unlikely that a trait contributes to the changes in the sleep EEG and in GH release. However, physiological sleep regulation, in which the balance of GHRH and CRH plays a key role, is clearly disturbed after TBI in previously normal young men.
Several other mechanisms, either alone or together, may contribute to these sequelae of TBI: 1) diffuse axonal injury (DAI); 2) elevated activity of the HPA system during stress exposure; and 3) therapeutic administration of glucocorticoids. DAI is the neuropathological correlate of damage in nearly every case of severe TBI.
35 Besides attention deficits and memory impairment, DAI may cause pathological changes in neurobiological regulating systems, such as sleep regulation via hypothalamic-releasing hormones, and it may even lead to decimation of GHRH-producing neurons. It is noteworthy in this connection that hypothalamic lesions following closed head injury have been described.
36 A diffuse hypothalamic injury could explain the blunted GH secretion and also the decrease in non-REM sleep.
Intensive care after TBI is accompanied by stress and hence by elevated CRH (and AVP) activity,
6 which also plays a key role in depression.
37 Accordingly, nonsuppression of serum cortisol after exogenous corticosteroids was reported in patients after TBI,
5,6 similar to nonsuppression after the dexamethasone suppression test in depression.
38 Parallel to our present findings, during and even after chronic stress in male rats there was shortened REM sleep latency, which is a sign of REM sleep disinhibition.
39 Furthermore, women in the process of getting a divorce who did not develop major depression showed REM disinhibition, and REM latency was shortened in a follow-up evaluation 1 to 2 years later.
40We should also note the possible role of corticosteroid treatment for intracranial pressure, which is often used in an acute intensive care situation after TBI. After acute administration of cortisol in normal control subjects, GH secretion and the amount of SWS increase,
41 but after chronic exogenous and endogenous hypercortisolism the opposite happens: there is decreased GH secretion and poor sleep continuity.
42 This mechanism of sleep-endocrine deterioration cannot be excluded in our sample because at least 4 patients had been treated with steroids several months before the study.
In summary, in this first investigation of sleep-endocrine alterations several months after severe TBI, we found a pattern of sleep-EEG parameters and nocturnal hormone secretion similar to that seen in patients with remitted depression. HPA overdrive and long-term modulation of hypothalamic and pituitary receptors may lead to permanent sleep-endocrine alterations, a neurobiological “scar.” An alternative, or perhaps additional, explanation is that there is hypothalamic-pituitary damage due to diffuse thinning out of neurons, such as GHRH-secreting cells.
Further studies, for example during administration of GHRH or GH-releasing peptides, might help elucidate the interactions between sleep and hormone secretion after TBI. In addition, a better understanding of the mechanisms of pathological changes after TBI might help in protecting the patients from subsequent depression. Early augmentational treatment with antidepressive drugs may be useful in the rehabilitation of patients after severe TBI. This issue appears to be an important one, considering that in a group of 66 patients hospitalized after TBI, Jorge and co-workers
25 found 28 (42%) who met the criteria for major depression.